Are meloxicam dimers really the structure-forming units in the ‘meloxicam–carboxylic acid’ co-crystals family? Relation between crystal structures and dissolution behaviour†
Nikolay A.
Tumanov
ab,
Svetlana A.
Myz
ab,
Tatyana P.
Shakhtshneider
ab and
Elena V.
Boldyreva
*ab
aInstitute of Solid State Chemistry and Mechanochemistry SB RAS, Novosibirsk, Russia. E-mail: eboldyreva@yahoo.com
bREC-008 Novosibirsk State University, Novosibirsk, Russia
First published on 25th October 2011
Abstract
We compare the packing of meloxicam in all the meloxicam-containing crystal structures known up to now, with a special emphasis on meloxicam and its co-crystals with carboxylic acids, two of which, with adipic and terephthalic acids, have not been reported before. We argue that it is not the meloxicam dimers, as was claimed in Cheney et al., Cryst. Growth Des. 2010, 10, 4401–4413, but a fragment containing two molecules of meloxicam linked via a carboxylic acid molecule that is the primary structure-forming element in all the known 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 meloxicam:carboxylic acid co-crystals. Meloxicam molecules form H-bonded dimers in the crystals of meloxicam, but these dimers are no longer present in any of the known meloxicam co-crystals. The molecules of meloxicam in some of its co-crystal structures can be occasionally close to each other as a consequence of a certain steric relation defined by the size of the carboxylic acid molecules. However, these molecular pairs termed “dimers” by Cheney et al. are different from the dimers in the crystals of meloxicam and can be held together by weak H-bonds only (if any), therefore they can hardly be considered as a structure-forming unit. An improved dissolution of meloxicam co-crystals as compared to poorly soluble meloxicam is supposed to be related to the presence of meloxicam dimers linked by relatively strong H-bonds in the crystals of meloxicam and to the absence of these dimers in its co-crystals.
:1 meloxicam:carboxylic acid co-crystals. Meloxicam molecules form H-bonded dimers in the crystals of meloxicam, but these dimers are no longer present in any of the known meloxicam co-crystals. The molecules of meloxicam in some of its co-crystal structures can be occasionally close to each other as a consequence of a certain steric relation defined by the size of the carboxylic acid molecules. However, these molecular pairs termed “dimers” by Cheney et al. are different from the dimers in the crystals of meloxicam and can be held together by weak H-bonds only (if any), therefore they can hardly be considered as a structure-forming unit. An improved dissolution of meloxicam co-crystals as compared to poorly soluble meloxicam is supposed to be related to the presence of meloxicam dimers linked by relatively strong H-bonds in the crystals of meloxicam and to the absence of these dimers in its co-crystals.
1. Introduction
Obtaining co-crystals of active pharmaceutical ingredients (APIs) with other small organic molecules is considered an important option in designing new drug forms.1 Apart from the aspect of patent protection,1c,2 introducing a drug molecule in a crystal structure with another component makes it possible to improve the properties as compared with those of the individual phases of drug crystals: making the form suitable for direct compression,3 less hygroscopic,1c,4 or increasing the release of the API into biological fluids on dissolution.1b,c,4b,c,5Crystal engineering is closely related to the problem of obtaining pharmaceutical co-crystals. First, it is important to be able to find a relation between crystal structure and properties in order to know which structures are required for achieving a required property (better compressibility, a higher dissolution rate, a requested melting temperature, etc.). Second, it is necessary to learn how to produce desirable structures when one of the molecular components (API) is fixed, but the other can be varied, e.g. different carboxylic acids can be selected. One of the methods of approaching these problems is to analyze a series of crystal structures trying to find structural fragments which are common for different crystals with similar properties.
Poor aqueous solubility is an industry-wide problem in drug discovery and the predominant problem associated with developing orally active drugs.1b,5f,6 80% of drugs are sold as tablets, 40% of drugs at the market have a solubility problem and 80% of drug candidates are poorly soluble. In general, it is obvious that the dissolution profile results from a complex interplay of the interactions between: i) molecules inside a crystal, ii) solvent molecules and the crystal-forming molecules at the crystal faces, and iii) molecules of the dissolved crystal and their solvation shell in solution. It can be therefore assumed that modifying intermolecular interactions in the crystal and making the interactions between the solute and the solvent molecules competitive with those between the molecules in the crystal may have a positive effect on the solubility. Co-crystal formation is one of the possible ways to modify the intermolecular interactions in the crystal. In co-crystals formed by two components A and B, some (or all) of the homomolecular A–A and B–B interactions are substituted for the heteromolecular A–B interactions, and this accounts for a change in the interaction with the solvent molecules on dissolution. One can suppose that, other parameters being similar, a co-crystal will dissolve easier as compared with the individual components if the heteromolecular interactions in the crystal are weaker than the homomolecular ones, or if the molecules in the co-crystals can be attacked easier by the solvent molecules on dissolution. An analysis of structure-forming fragments in a series of co-crystals and of the individual compound in a comparison with their dissolution kinetics may help to understand the relative role of different intermolecular interactions, both in forming crystal structures (‘crystal engineering’) and in the dissolution.
Meloxicam, 4-hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxide (Fig. 1) is becoming popular as a non-steroidal anti-inflammatory drug which combines high therapeutic efficacy with much lower levels of side-effects.7 Unfortunately, meloxicam is poorly soluble in water, and this decreases its bioavailability. Many recent publications deal with the problem of solubilization of meloxicam. The main methods of increasing the dissolution rate or the solubility of this compound are related to preparing complexes with β-cyclodextrine,8 formulations with polymeric excipients,9 dispersions with porous minerals, such as calcium silicate,10 salts,11 and micronized or nano-size particles.12
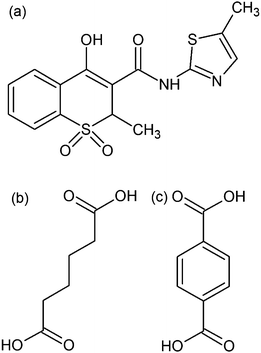 | ||
| Fig. 1 Molecular schemes of meloxicam in the enol prototropic form (a), adipic acid (b) and terephthalic acid (c). | ||
Co-crystals of meloxicam with carboxylic acids were also shown to have higher solubility and dissolution rate, and thus to be promising for pharmaceutical applications.13 The reason for their improved dissolution profiles remains unclear. In the crystal structure of meloxicam, molecules form dimers linked via N–H⋯O(=S) H-bonds (Fig. 2).14 One could expect the increased dissolution rate of meloxicam co-crystals as compared to meloxicam to be related to breaking these dimers and thus making meloxicam molecules more accessible for interactions with solvent in aqueous solutions. In a recent paper15meloxicam dimers were claimed to be the basic structure-forming unit in co-crystals of meloxicam with carboxylic acids: “It is observed that the meloxicam dimer is robust since this motif is observed in five out of six meloxicam co-crystal structures that have been determined”.15 We have analyzed the structures of co-crystals described and have noticed that they have no dimers, which are present in meloxicam itself, but only molecular pairs, characterized by rather long intermolecular O–H⋯O contacts (O–O distance is about 2.872–3.055 Å) with O–H–O angles about 110–115°, which can be classified as weak H-bonds only (the same molecular pairs are present also in the meloxicam crystals, in addition to the dimers bonded by stronger NH⋯O bonds). The aim of the present work was to revisit the problem of the presence of meloxicam dimers in the crystals of meloxicam, in its salts and hydrate, as well as in meloxicam co-crystals with carboxylic acids, including some new structures not described earlier, and to consider crystal structures in relation to the dissolution behaviour.
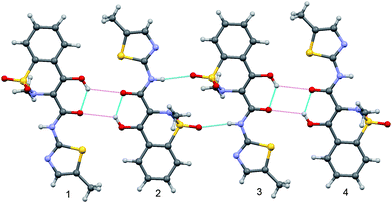 | ||
| Fig. 2 A fragment of meloxicam structure showing a meloxicam dimer linked by NH⋯O hydrogen bonds (molecules 2–3) and two ‘molecular pairs’ (molecules 1–2 and 3–4. NH⋯O intermolecular and OH⋯O intramolecular hydrogen bonds are shown by blue lines, O–H⋯O contacts with longer O–O distances between molecules in ‘molecular pairs’ are shown as weaker magenta dashed lines. | ||
2. Materials and methods
Samples
Meloxicam was purchased from Chem-East Ltd., Hungary, carboxylic acids from Hebei Welcome Pharmaceutical Co., Ltd, China and solvents from Reakhim, Russia and Sigma-Aldrich, USA. Three techniques were used for the synthesis of co-crystals of meloxicam with 17 carboxylic acids (in 2:1 ratio‡): co-crystallization from solutions in different solvents (THF, toluene, ethanol, benzene, dioxane), heating of dry mixtures and solvent-drop co-grinding in a mortar or in a mill (SPEX 8000, CertiPrep Inc., USA). Details of synthesis have been described elsewhere.16
X-ray single-crystal diffraction and crystal structure analysis
Diffraction experiments were carried out using an Oxford Diffraction Gemini Ultra R CCD diffractometer (graphite monochromator, Mo-Kα; λ = 0.7173 Å, 293 K). For new meloxicam–adipic acid (1) and meloxicam–terephthalic acid (2) co-crystals the structures were solved by direct methods using SHELXS9717 and refined by full matrix least-squares based on F2 using SHELXL97.17 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were located in the difference Fourier map. For (1), some hydrogen atoms were refined freely and some as riding atoms with isotropic parameters 1.2 times those of the heavy atoms to which they are attached. Hydrogen atoms in (2) were refined freely. CIFs are available as ESI (Crystal structure data†) and deposited with the Cambridge Structural Database (CCDC reference numbers 834808 and 834809 for (1), (2), respectively). A summary of crystal data, structure solution and refinement is also available as an ESI file (Supplementary Information, ESI-1).† Platon, Mercury and Crystal Explorer were used for the analysis of crystal structures.18CSD analysis using materials module in mercury
Crystal packing was analyzed using the ‘Crystal Packing Similarity’ procedure in the Materials module of Mercury CSD 2.4.5. A set of 20 structures (meloxicam14§, a hydrate,14potassium and hydrosulphate hydrates,14,20 metal complexes,21meloxicam bromide,22 and co-crystals with carboxylic acids13,15,this work) was used in analysis. This set includes, to the best of our knowledge, all known crystal structures containing meloxicam in different propionic forms. Structural data were taken from the original publications; some structures were retrieved from the CSD version 5.3223 with updates up to May 2011 (REFCODEs: CAKLOQ, CAKLUW, CAKMAD, CIHZOJ, SEDZOQ01, UHIMON, WODBEW, WODBIA, WODBOG, LAKKOZ).The tree diagram was generated using the ‘Crystal Packing Similarity’ tool as described in the Mercury tutorial.18b,c,24 In metal-containing compounds, metal atoms were deleted in a few CIFs, to make the meloxicam fragment ‘free’, to enable a comparison with organic structures. The default values for distance (20%), and angle tolerance (20°) were used. The ‘ignore number of hydrogens’, ‘ignore number of bonded atoms’, ‘ignore each atom's hydrogen counts’, ‘ignore each atom's bond counts’, ‘ignore smallest molecular components’ options were used. The search was performed for each level of cluster size from 15 to 2 molecules, but levels without any changes were omitted on the tree diagram. Results of these searches were used to plot the connection in the branched tree diagram, which illustrated similarity of structures.
IR spectroscopy
Attenuated total reflectance (ATR) IR spectra were measured without any special sample preparation in the spectral range of 4000–580 cm−1 using an FTIR spectrometer Digilab Excalibur 3100 (USA) and a Pike Miracle ATR attachment with a ZnSe crystal.Dissolution tests
Dissolution of meloxicam and its co-crystals in 200–250 ml of phosphate buffer solution with pH = 6.8 at 37 ± 0.5 °C was studied using a Varian 705 DS dissolution tester and a Cary 50 optical spectrometer following the intensity of the 362 nm band in the spectrum of meloxicam. Powder samples were sieved prior to dissolution tests, and the fractions with particle size 315–125 μm were selected.3. Results and discussion
In the only known crystal structure of meloxicam, molecules form dimers linked via N–H⋯O hydrogen bonds (Fig. 2). The geometrical parameters of these N–H⋯O bonds (D⋯A = 3.035(3) Å, H⋯A = 2.24(4) Å, D–H⋯A = 167(3)°)14 are typical for medium H-bonds.25 There is also a relatively strong intramolecular O–H⋯O H-bond (D⋯A = 2.574(3) Å). Oxygen atoms of the neighbouring molecules are too far from each other to qualify the intermolecular O–H⋯O contacts as H-bonds according to commonly accepted criteria (D⋯A = 3.236(3) Å, O–H–O angle equal to 129(4)°). Still, the same pattern in meloxicam co-crystals is termed in ref. 15 as a ‘dimer’, which is claimed to be held “via electrostatic interactions between the alcohol, ketone, and sulfathiazole moieties”. We shall term the pattern further on as a ‘molecular pair’, to distinguish it from the dimer in meloxicam crystals, which is held by N–H⋯O bonds.In all the 2:1 meloxicam co-crystals with carboxylic acids, meloxicam molecules are present in the enol prototropic form, the same as in the structure of meloxicam itself. Molecular conformation is stabilized by an intramolecular O–H⋯O hydrogen bond, which is preserved in all the structures. The intramolecular bond lengths and angles, as well as the length of the intramolecular H-bond, are similar in the structure of meloxicam, and in all the meloxicam co-crystals. The most remarkable difference in the intramolecular geometry is related to the values of the angle between the planes of the thiazole and benzene rings. The values of 24.0 and 20.2° in co-crystals with adipic and terephthalic acids, respectively, are the largest for all the known meloxicam-containing structures, with the only exception of meloxicam hydrosulphate hydrate, in which this value is even larger (26.6°).14 For a comparison, the value of this angle in the structure of meloxicam–succinic acid co-crystal is equal to 5.5°,13,15 and is similar to that in other meloxicam co-crystals.
No meloxicam dimers linked via N–H⋯O hydrogen bonds are present in its co-crystals. Meloxicam ‘molecular pairs’ (termed ‘dimers’ in ref. 15) (Fig. 3a) can be found in the structures of meloxicam co-crystals with 1-hydroxy-2-naphthoic, glutaric, L-malic, fumaric and succinic acids.15 In the structure of meloxicam–salicylic acid co-crystal the neighbouring meloxicam molecules are approximately perpendicular to each other, which makes the formation of ‘molecular pairs’ impossible. In the structures of meloxicam–adipic acid and meloxicam–terephthalic acid co-crystals one can notice ‘chains’ with short S–O contacts (3.280(2)/3.088(1) Å) (Fig. 3b) instead of ‘molecular pairs’. Similar ‘chain fragments’ can be found in the crystal structures of meloxicam hydrosulphate monohydrate14 and in the benzene solvate of trans-dichloro-(η2-ethylene)-(meloxicam)-Pt(II).21b A more careful examination of the structures of meloxicam co-crystals with carboxylic acids (2:1) reveals much similarity between all of them, independently of the presence or the absence of the ‘molecular pairs’ noticed by Cheney et al.,15 or ‘chains’. Taking into account that no strong directional specific intermolecular interactions can be related either to ‘molecular pairs’ or to ‘chains’, one can suppose that these motifs are formed virtually due to steric effects, and can hardly be considered as structure-forming units, or synthones.
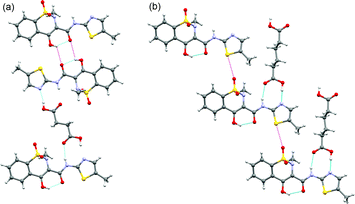 | ||
| Fig. 3 Structural fragments of the co-crystals of meloxicam with succinic acid (a) and adipic acid (b) showing, respectively, the ‘molecular pairs’ and ‘chain fragments’ packing motifs. Short contacts and O–H⋯O contacts with long O–O distances, not qualified as H-bonds, are shown as magenta dashed lines. | ||
In the structures of meloxicam co-crystals with carboxylic acids, homomolecular contacts are substituted for heteromolecular ones, and the H-bonded meloxicam dimers are not preserved. Typical linkage between a meloxicam molecule and a carboxylic acid molecule is the same in all the co-crystal structures: the carboxylic group of an acid forms N–H⋯O and O–H⋯N hydrogen bonds with NH- and thiazole-groups of meloxicam, respectively. In all the (2:1) meloxicam co-crystals known up to now one can find the same structural fragment containing two molecules of meloxicam linked via a carboxylic acid molecule (Fig. 4).¶ Surprisingly, a very similar motif can be found also in the structure of meloxicam hydrogen sulphate monohydrate (Fig. 4d): two water molecules and two hydrogen sulphate anions form a ‘structural analogue’ of a terephthalic acid molecule, so that not only the topology of the intermolecular contacts, but also the interatomic distances in the structures are similar (Table 1). These structural motifs are packed in flat layers (Fig. 5), which can also be found in all the known meloxicam co-crystals with 2:1 stoichiometry. (See ESI-3†). In the structures of the co-crystals with adipic and terephthalic acids the distance between two meloxicam molecules linked via a carboxylic acid molecule is large enough to partly incorporate a meloxicam molecule from a neighbouring ‘2:1 structural fragment’. This prevents the formation of ‘molecular pairs’, which were selected in ref. 15 as a structure-forming synthone in other co-crystals, where the distance between the meloxicam molecules in a ‘2:1 synthone’ is not sufficient to incorporate one more molecule (Fig. 5).
| Adipic acid | Terephthalic acid | Hydrosulphate monohydrate | Succinic acid | |
|---|---|---|---|---|
| a For meloxicam hydrogen sulphate monohydrate. | ||||
| N–N | 12.681(3) | 12.460(2) | 12.786(4) | 10.403(2) |
| O–O | 7.355(3) | 6.956(2) | 7.463(3) | 5.023(2) |
| D–A (Nthiazole⋯O–H) | 2.663(3) | 2.639(2) | 2.662(5) | 2.697(2) |
| D–A (N–H⋯O /N⋯H–Oa) | 2.866(3) | 2.984(2) | 2.798(4) | 2.871(2) |
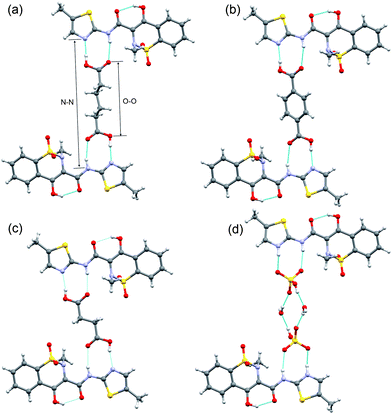 | ||
| Fig. 4 Structure-forming fragments of meloxicam co-crystals with adipic (a), terephthalic (b), succinic (c) acids, and meloxicam hydrogen sulphate monohydrate (d). Hydrogen bonds are shown by blue lines. N–N and O–O distances are shown by arrows. Similar structure-forming fragments for other co-crystals are given in ESI-2.† | ||
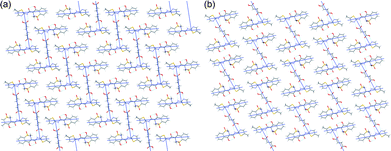 | ||
| Fig. 5 Packing of the ‘structure-forming fragments’ in the meloxicam co-crystals with adipic (a) and succinic (b) acids. Similar packing motifs for other co-crystals are given in ESI-3.† | ||
The differences in the molecular packing within a layer manifest themselves in the distribution of contacts of meloxicam molecules with its nearest environment in the structure. A mere visual inspection of the graphical representations of Hirshfeld surfaces18d–f does not seem to reveal any significant differences (Fig. 6, ESI-4†), thus supporting the conclusion that all the meloxicam co-crystals are structurally related. However, a more detailed analysis of the distribution of contacts sorted according to atom types (Table 2) reveals a minor difference between the crystal structures with meloxicam ‘molecular pairs’ and meloxicam ‘chains’. The difference in the meloxicam packing is seen better not by the distribution of contacts with the largest contributions (H–H, O–H, C–H), but when relatively small contributions (C–C, C–N, C–S, C–O, S–H, S–O, O–O) are considered. The first types of contacts depend mainly on the kind of co-former, whereas the second depend on the meloxicam–meloxicam contacts. The most pronounced difference in the Hirshfield fingerprint plots of different structures (a smeared long-range distances region) is related, mainly, to the contributions of the C–H and H–H contacts. It is better seen with the decomposed Hirshfield fingerprint plots, which are given in ESI-5.† The contribution of these contacts depends mainly on the type of the carboxylic acid, and not on the mutual juxtaposition of meloxicam molecules; therefore these numbers do not allow one to distinguish between two packing ‘types’. Since all the crystal structures also have the same fragment containing N–H⋯O and O–H⋯N hydrogen bonds, the contribution of the N–H contacts is also approximately the same in all the structures, and this parameter cannot be used to distinguish between the packing ‘types’. Besides, the same structural fragments give a similar contribution of the O–H contacts. The juxtaposition of meloxicam molecules with respect to each other depends on the contributions of C–C, C–N, C–S, C–O, S–H, S–O, O–O contacts, and, partly, also of the O–H contacts. The analysis of these values allows one to reveal the differences between two packing ‘types’ (Table 2).
| 1 | 2 | 7 | 3 | 4 | 5 a | 5 b | |
|---|---|---|---|---|---|---|---|
| a Meloxicam in the enol form. b Meloxicam in the cation form. | |||||||
| C–H,% | 11.0 | 16.5 | 9.2 | 8.9 | 8.2 | 9.0 | 9.8 |
| C–C,% | 0.5 | 0.8 | 0.3 | 3.4 | 4.3 | 3.1 | 3.2 |
| C–N,% | 0.8 | 0.2 | 0.4 | 1.8 | 1.8 | 1.6 | 1.4 |
| C–S,% | 3.3 | 3.1 | 3.0 | 1.6 | 1.6 | 1.6 | 1.6 |
| C–O,% | 6.0 | 5.4 | 8.5 | 2.9 | 3.4 | 3.2 | 3.4 |
| H–H,% | 35.5 | 29.4 | 30.0 | 33.8 | 32.8 | 32.0 | 32.8 |
| N–H,% | 5.7 | 6.2 | 3.2 | 4.9 | 5.1 | 5.3 | 2.5 |
| S–H,% | 2.9 | 3.2 | 2.8 | 4.9 | 5.0 | 4.8 | 4.8 |
| O–H,% | 30.1 | 30.8 | 38.7 | 32.9 | 32.9 | 34.1 | 35.8 |
| N–O,% | 0.7 | 0.5 | 0.9 | 0.7 | 0.7 | 0.6 | 0.6 |
| S–O,% | 2.3 | 2.5 | 1.5 | 1.2 | 1.2 | 1.3 | 1.3 |
| S–S,% | — | — | — | 0.7 | 0.8 | 0.7 | 0.7 |
| S–N,% | — | — | — | 0.3 | 0.3 | 0.3 | 0.3 |
| O–O,% | 1.3 | 1.3 | 1.5 | 2.1 | 2.1 | 2.2 | 1.9 |
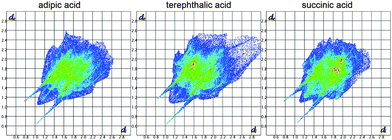 | ||
| Fig. 6 Two-dimensional Hirshfield fingerprint plots of structures of meloxicam co-crystals with adipic, terephthalic and succinic acids. The colour in the sequence white–blue–green–red is a summary of the frequencies of each combination of distances de and di across the Hirshfield surface of a molecule (in increasing order), where di is internal distance and de external distance from the Hirshfeld surface to the nearest molecule. Long ‘wings’ correspond to N–H⋯O and O–H⋯N hydrogen bonds. | ||
Analysis of meloxicam packing using a similarity tree diagram18b,c,24 (Fig. 7) also helps to reveal differences and similarities between various meloxicam-containing crystal structures. This technique allows one to calculate the level of packing similarity between structures containing the same compound using the geometry of a cluster of selected molecules around a reference molecule to determine the number of similar molecules in the cluster and the root-mean-squared deviation (RMSD) in atomic positions of overlapped molecules. The more overlapped molecules in cluster, the more similar the structures are. Overlapping of 15–16 molecules in the cluster and a low RMSD (less than ∼1 Å) means that compared structures are virtually isostructural. Co-crystals of meloxicam with adipic, terephthalic acids and with hydrogen sulphate monohydrate form one similarity group, with 15 molecules in coinciding clusters, which means practically complete isostructurality in meloxicam packing. If the number of molecules in the coinciding cluster is reduced to two, two more structures are added to this group. This should mean that although the structures in a new (extended) similarity group are quite different, there is similarity in the structures of small fragments. Another group is formed by meloxicam co-crystals with succinic, fumaric and L-malic acids; if the number of molecules in the coinciding clusters is reduced to 14, a co-crystal of meloxicam with glutaric acid (1:1) is also added to this group, despite the difference in the stoichiometry. The tree diagram also shows that this group has a small similar fragment with the structures of meloxicam and of its co-crystal with 1-hydroxy-2-naphthoic acid. All the other structures are far on the tree diagram from the structures of meloxicam and meloxicam co-crystals, i.e. have no common structural motifs with them. Examples of clusters, which are similar in different structures corresponding to selected similarity groups, are shown in the enlarged version of Fig. 7 given in ESI-6.†
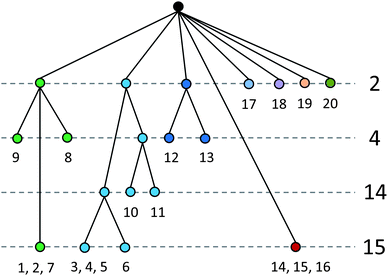 | ||
| Fig. 7 A tree diagram based on the results from the ‘Crystal Packing Similarity’ tool in the Materials module of Mercury. 1, meloxicam–adipic acid co-crystal, 2, meloxicam–terephthalic acid co-crystal, 3, meloxicam–succinic acid co-crystal, 4, meloxicam–fumaric acid co-crystal, 5, meloxicam-L-malic acid co-crystal, 6, meloxicam–glutaric acid co-crystal, 7, meloxicam hydrogen sulphate monohydrate, 8, trans-dichloro-(H2-ethylene)-(meloxicam)-platinum(II) benzene solvate, 9, meloxicam bromide, 10, meloxicam, 11, meloxicam-1-hydroxy-2-naphthoic acid co-crystal, 12, potassium salt monohydrate of meloxicam, 13, cis-diamine-bis(meloxicamato)-platinum(II) dimethylsulfoxide solvate tetrahydrate, 14, trans-dimeloxicam-trans-bis(dimethylsulfoxide)-cobalt(II), 15, trans-dimeloxicam-trans-bis(dimethylsulfoxide)-zinc(II), 16, trans-dimeloxicam-trans-bis(dimethylsulfoxide)-cadmium(II), 17, ammonium meloxicam, 18, meloxicam hydrate, 19, bis(meloxicamato)-(dimethylformamide)-copper(II) hydrate, 20, meloxicam–salicylic acid co-crystal form III. Structures grouped (e.g., 1, 2, 7) in the bottom row indicate that 15 out of 15 molecules in a cluster are similar, i.e. these crystals are considered to be isostructural. See also ESI-6.† | ||
All meloxicam co-crystals with carboxylic acids show an increase in the dissolution rate in comparison with meloxicam.16 As an example, the dissolution kinetics of meloxicam and its co-crystals with succinic, adipic and terephthalic acids are compared in Fig. 8. The supersaturated solution is formed at the initial stage of dissolution of co-crystals, and the supersaturation is preserved for hours. Such a behaviour is especially favourable for pharmaceutical applications.6 This effect is the most pronounced for meloxicam–terephthalic acid, for which it significantly exceeds the increase in the dissolution rate and apparent solubility, as compared not only with commercial meloxicam, but also with the more soluble meloxicam samples subjected to mechanical treatment.16 At the same time, the difference in the dissolution of meloxicam–adipic and meloxicam–succinic acid is noticeably smaller. The results seem to support the hypothesis, which has motivated the present study: breaking true H-bonded meloxicam dimers and substituting homomolecular contacts for heteromolecular ones does facilitate the dissolution of meloxicam co-crystals as compared with meloxicam. At the same time, the presence or the absence of ‘molecular pairs’ seems not to have a pronounced effect on the dissolution behaviour, supporting the hypothesis that the interactions between the molecules in ‘molecular pairs’ are weak.||IR spectra of meloxicam co-crystals with different carboxylic acids (Fig. 9) also do not reveal any significant differences, which could indicate noticeable differences in the intermolecular interactions in the crystal structures with ‘chains’ and with ‘molecular pairs’. At the same time, the IR spectra of meloxicam co-crystals differ from those of meloxicam. The strong ν(N–H) peak of meloxicam at 3290 cm−1 is shifted to lower frequencies sharply decreasing in peak intensity, although the integral intensity is preserved or even increases. The ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching mode, νas(SO2) mode and ν(C–N) + δ(NH) combination and amide III band are shifted to higher frequencies. These changes correspond to the strengthening of the NH⋯O hydrogen bonds, as homomolecular dimers in meloxicam are substituted for the heteromolecular meloxicam–carboxylic acid trimers in co-crystals.
O) stretching mode, νas(SO2) mode and ν(C–N) + δ(NH) combination and amide III band are shifted to higher frequencies. These changes correspond to the strengthening of the NH⋯O hydrogen bonds, as homomolecular dimers in meloxicam are substituted for the heteromolecular meloxicam–carboxylic acid trimers in co-crystals.
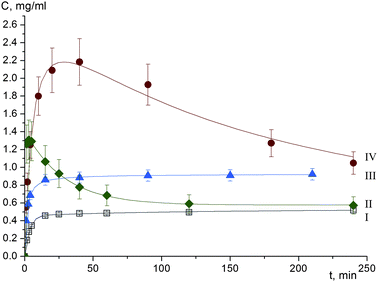 | ||
| Fig. 8 Dissolution profiles of meloxicam (I), and co-crystals of meloxicam with succinic (II), adipic (III), terephthalic (IV) acids. | ||
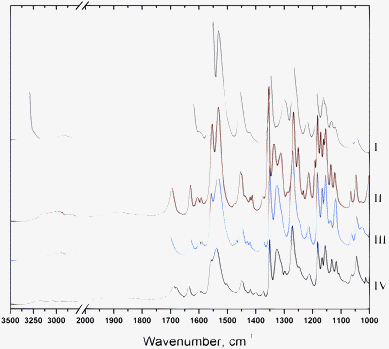 | ||
| Fig. 9 IR spectra of meloxicam (I) and co-crystals of meloxicam with succinic (II), adipic (III), terephthalic (IV) acids. | ||
The solubility problem is one of the major problems of solid dosage forms development.1b,5f,6 Substituting homomolecular contacts in a crystal for heteromolecular ones (for example, by preparing co-crystals) may be one of the ways of facilitating the dissolution. This effect can be observed both if homomolecular interactions are stronger than the heteromolecular ones, and if this substitution facilitates interaction between the molecules in the crystal and the solvent, e.g. because of steric reasons.
In general, the lattice energy and the energy of interactions in a crystal should not necessarily correlate with solubility. For example, the crystals of NaCl are very well soluble in water, although the lattice energy is high, and very high temperatures are needed for melting. Some co-crystals of meloxicam have slightly lower melting temperatures than meloxicam, and others have slightly higher,15,16 but all are soluble faster, and there is no correlation between the melting temperature and the dissolution profile, if different co-crystals are compared with each other.16 For example, meloxicam melts at 240 °C, its co-crystals with adipic acid at 195 °C, with terephthalic acid at 255 °C, and with succinic acid at 230 °C.
In case of organic co-crystals, one can imagine two situations depending on whether the crystallization of a co-crystal from a multi-component solution corresponds to the formation of the equilibrium, or of the non-equilibrium phase. Heteromolecular interactions can be stronger, than the homomolecular ones, and the stability of the co-crystal higher than that of the individual components. Alternatively, heteromolecular interactions in a co-crystal can be weaker, as compared to the homomolecular interactions in the phases of the individual components. This does not contradict the very possibility for the co-crystal to be formed. When several solid phases compete for crystallization in a system, it is not necessarily the most stable one which is formed, but the one which forms faster. This has been known for over a century for single-component crystals as Ostwald's rule of stages.27 There is also experimental evidence that co-crystals are often formed as metastable phases upon rapid crystallization, whereas individual components crystallize from the same solution upon slow solvent evaporation. This may explain why co-crystals are often obtained by such techniques as anti-solvent crystallization, spray-drying, or mechanical co-grinding. This may also explain why different polymorphs of a co-crystal with the same composition are easily formed under different conditions.28
Obviously enough, breaking H-bonded homomolecular dimers in a co-crystal of an API can be expected to facilitate its dissolution when A–B interactions are weaker than A–A and B–B interactions. At the same time, even if the lattice energy of a co-crystal is higher than that of the individual components (because of other interactions than H-bonds in the homomolecular dimers), the co-crystal can be dissolved easier in an aqueous solution: the contact of the API molecules in the crystal with water can be facilitated if the H-bonds between the molecules of API are broken, e.g. because of steric reasons. In meloxicam there are real dimers with NH⋯O hydrogen bonds, and these real dimers are broken when a co-crystal is formed. New NH⋯O bonds between meloxicam and carboxylic acid molecules are formed, and they can be even stronger, than the bonds in homomolecular dimers in meloxicam were, as can be supposed based on the IR spectra and the N–O distances from diffraction data (3.035 Å in meloxicam, 2.984–2.866 Å in co-crystals). However, even in this case the interactions with the solvent may be facilitated and the meloxicam molecules more accessible for an “attack” in a “carboxylic acid–meloxicam trimer”, than when meloxicam molecules form centrosymmetric homomolecular dimers. Interactions other than H-bonds (aromatic interactions, or van der Waals interactions contributing to packing coefficients) may be important for the total lattice energy, thermal stability or melting, but apparently not for the dissolution in water.
Conclusion
The present work has shown that an analysis of the structure-forming fragments in a series of co-crystals and of the individual compound in a comparison with their dissolution kinetics may help to understand the relative role of different intermolecular interactions, both in forming crystal structures (‘crystal engineering’) and in the dissolution kinetics. A similar comparison of the dissolution behaviour of individual phases with that of a series of co-crystals, with a special focus on the existence (or the absence) of a correlation between breaking H-bonded homomolecular dimers in the co-crystal structure, could be extended from meloxicam to other systems, in which molecules form H-bonded dimers in the crystals of individual compounds, such as, e.g., aspirin,29indomethacin,30piroxicam,31α-glycine.32 For some of these compounds, also polymorphs not containing H-bonded dimers, as well as co-crystals preserving or not the H-bonded dimers (see ref. 4b, 28b, 28o and 33, as examples) are known. To the best of our knowledge, no attempts to consider systematically a correlation between breaking the dimers and improving the dissolution behaviour have been made, although a brief preliminary analysis of the data already available suggests that this correlation might exist not for meloxicam co-crystals only. For example, in contrast to the stable γ-polymorph of indomethacine,30a the H-bonded homomolecular dimers are no longer present in the co-crystals with lidocaine,33a,b and are partly broken in the α- polymorph30d – these forms are more soluble as compared with γ-polymorph. Homomolecular dimers are broken in a co-crystal of glutaric acid with 2-[4-(4-chloro-2-fluorophenoxy)phenyl]pyrimidine-4-carboxamide, which shows a significant improvement in the dissolution profile and in pharmocokinetics.5d It would be most interesting to compare the dissolution of different co-crystals of piroxicam (a therapeutic and a structural analogue of meloxicam) with carboxylic acids, since in some of them homomolecular dimers are preserved, whereas in other they are not,33c and it is known that the more soluble34 α-polymorph33g of piroxicam has no H-bonded dimers, whereas β-polymorph33h has. Unfortunately, no data for the dissolution kinetics of piroxicam co-crystals seems to be available. A series of various pharmaceutical co-crystals was designed recently in order to insert a complementary molecular component into the hydrogen-bond motif of the parent crystal, and to observe the effect on the intermolecular interactions in the interlayer region. The designed co-crystals were successfully produced either by solution crystallization, or by co-grinding, but no data on the dissolution behaviour of these co-crystals as compared to individual components have been reported.28o A systematic comparison of the dissolution of co-crystals in relation to the presence/absence of homomolecular/heteromolecular bonds and their relative energies would complement the existing approaches to the problem of co-crystal dissolution, which consider the packing density,35 co-crystal eutectic constants,36 or the effect of a co-former on pH, complex or micelle formation in solution.37 This would be a step forward in designing more soluble structures, as compared to much simpler (though still giving some right predictions) approaches like counting the number of hydrogen bonds per drug molecule and deriving ‘rules of thumbs’ out of these calculations.6Acknowledgements
This work was supported by grants from CRDF (RUX0-008-NO-06), RF Ministry of Education and Science (2.2.2.2/10470), RFBR (10-03-00252, 11-03-00684), Council for Grants of the President of RF for Support of Leading Scientific Schools (NSh-4357.2010.3), Projects 5.10, 21.44 and 5.6.4 of the Russian Academy of Sciences. The authors thank Dr V. A. Drebushchak for measuring the melting temperatures of the studied compounds and Dr Yu. A. Chesalov for helpful discussions of the IR spectra.Notes and references
- (a) S. L. Morissette, O. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef; (b) N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS; (c) N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CrossRef CAS; (d) W. Jones, W. D. S. Motherwell and A. V. Trask, MRS Bull., 2006, 31, 875–879 CAS; (e) P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef; (f) B. S. Sekhon, Ars Pharmaceutica, 2009, 50, 99–117 Search PubMed; (g) T. Friščić and W. Jones, J. Pharm. Pharmacol., 2010, 62, 1547–1559 CrossRef CAS.
- A. V. Trask, Mol. Pharmaceutics, 2007, 4, 301–309 CrossRef CAS.
- S. Karki, T. Friscic, L. Fabian, P. R. Laity, G. M. Day and W. Jones, Adv. Mater., 2009, 21, 3905–3509 CrossRef CAS.
- (a) A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CrossRef CAS; (b) S. Basavoju, D. Boström and S. P. Velaga, Pharm. Res., 2008, 25, 530–541 CrossRef CAS; (c) S. Cherukuvada, N. J. Babu and A. Nangia, J. Pharm. Sci., 2011, 100, 3233–3244 Search PubMed.
- (a) D. J. Good and N. Rodriguez-Hornedo, Cryst. Growth Des., 2010, 10, 1028–1032 CrossRef; (b) D. J. Good and R.-H. Naír, Cryst. Growth Des., 2009, 9, 2252–2264 CrossRef CAS; (c) D. P. McNamara, S. L. Childs, J. Giordano, A. Iarriccio, J. Cassidy, M. S. Shet, R. Mannion, E. O'Donnell and A. Park, Pharm. Res., 2006, 23, 1888–1897 CrossRef CAS; (d) Z. J. Li, Yu. Abramov, J. Bordner, J. Leonard, A. Medek and A. V. Trask, J. Am. Chem. Soc., 2006, 128, 8199–8210 CrossRef CAS; (e) K. Shiraki, N. Takata, R. Takano, Y. Hayashi and K. Terada, Pharm. Res., 2008, 25, 2581–2592 CrossRef CAS; (f) N. J. Babu and A. Nangia, Cryst. Growth Des., 2011 Search PubMed.
- C. Lipinski, Amer. Pharm. Rev., 2002, 5, 82–85 Search PubMed.
- M. Del Tacca, R. Colucci, M. Fornai and C. Blandizzi, Clin. Drug Investig., 2002, 22, 799–818 Search PubMed.
- (a) A. Obaidat and R. Obaidat, Acta Pharm., 2011, 61, 83–91 Search PubMed; (b) A. A. Obaidat, R. A. Khanfar and M. N. Khawam, J. Inclusion Phenom. Macrocyclic Chem., 2009, 63, 273–279 Search PubMed; (c) G. El-Mahrouk, M. H. Aboul-Einien and N. A. Elkasabgy, Asian J. Pharm. Sci., 2009, 4, 8–22 Search PubMed; (d) M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS.
- (a) S. V. Kulkarni, K. P. Ranjit, N. Patel, R. B. Someshwara, B. Ramesh and K. P. Ashok, Intern. J. Pharm. Pharm. Sci., 2011, 3, 91–93 Search PubMed; (b) S. Emmadi, K. Sanka, A. R. Potu, R. Jukanti, S. Bandari and P. R. Veerareddy, Latin Amer. J. Pharm., 2010, 29, 1303–1310 Search PubMed; (c) A. Niharika, K. Sundaramoorthy and T. Vetrichelvan, Res. J. Pharm., Biol. Chem. Sci., 2010, 1, 655–671 Search PubMed; (d) A. Aejaz, M. Jafar, M. H. G. Dehghan and S. Adil Shareef, Intern. J. Pharm. Pharm. Sci., 2010, 2, 182–190 Search PubMed; (e) M. M. Ghareeb, A. A. Abdulrasool, A. A. Hussein and M. I. Noordin, AAPS PharmSciTech, 2009, 10, 1206–1215 Search PubMed; (f) H. A. El-Maradny, S. A. Mortada, O. A. Kamel and A. H. Hikal, Acta Pharm., 2008, 58, 455–466 Search PubMed; (g) A. Bashiri-Shahroodi, P. R. Nassab, P. Szabó-Révész and R. Rajkó, Drug Dev. Ind. Pharm., 2008, 34, 781–788 Search PubMed; (h) M. A. Hassan, S.T.P. Pharma Pratiques, 2008, 18, 275–284 Search PubMed; (i) N. Inamdar, K. Bhise and S. Memon, Asian J. Pharm., 2008, 2, 128–132 Search PubMed; (j) D. Pathak, S. Dahiya and K. Pathak, Acta Pharm., 2008, 58, 99–110 Search PubMed; (k) M. El-Badry and M. Fathy, Drug Dev. Ind. Pharm., 2006, 32, 141–150 Search PubMed; (l) M. Hassan, G. Dehghan and M. Jafar, Iranian J. Pharm. Res, 2006, 4, 231–238 Search PubMed; (m) V. Kumar and D. N. Mishra, Yakugaku Zasshi, 2006, 126, 657–664 Search PubMed; (n) P. R. Nassab, R. Rajko and P. Szabo-Revesz, J. Pharm. Biomed. Anal., 2006, 41, 1191–1197 Search PubMed; (o) B. Naidu, K. P. Chowdary, K. V. Murthy, V. Satyanarayana, A. R. Hayman and G. Becket, J. Pharm. Biomed. Anal., 2004, 35, 75–86 CrossRef CAS; (p) K. P. R. Chowdary, Ind. J. Pharm. Sci., 2001, 63, 150–154 Search PubMed.
- (a) U. Mukhija, A. Bhargav and D. C. Bhatt, Intern. J. Pharm. Recent Res., 2010, 2, 69–75 Search PubMed; (b) S. Sharma and A. Pawar, Int. J. Pharm., 2006, 313, 150–158 Search PubMed; (c) M. Kinoshita, K. Baba and A. Nagayasu, J. Pharm. Sci., 2002, 91, 362–370 Search PubMed; (d) T. P. Shakhtshneider, S. A. Myz, N. I. Nizovskii, E. V. Boldyreva, T. C. Alex, Rakesh Kumar, Abstracts Intern. Confer. Chem. Organic Solid State (ICCOSS XX). Bangalore, India, June 26–30, 2011 Search PubMed; (e) V. P. Isupov, T. P. Shakhtshneider, S. A. Myz, V. V. Boldyrev, Russian Patent RF № 2421243, priority from 09.11.2009, Published on 20.06.2011, Bulletin № 17; (f) A. I. Nizovsky, A. V. Kalinkin, T. P. Shakhtshneider, M. A. Dyakonova, S. A. Myz, E. V. Boldyreva, Rakesh Kumar, J. Struct. Chem, in press Search PubMed.
- H.-K. Han and H.-K. Choi, Eur. J. Pharm. Biopharm., 2007, 65, 99–103 CrossRef CAS.
- (a) R. Ambrus, P. Kocbek, J. Kristl, R. Šibanc, R. Rajkó and P. Szabó-Révész, Int. J. Pharm., 2009, 381, 153–159 Search PubMed; (b) A. H.-J. Chiou, M.-K. Yeh, C.-Y. Chen and D.-P. Wang, J. Supercrit. Fluids, 2007, 42, 120–128 Search PubMed.
- (a) S. A. Myz, T. P. Shakhtshneider, K. Fucke, A. P. Fedotov, E. V. Boldyreva, V. V. Boldyrev and N. I. Kuleshova, Mendeleev Commun., 2009, 19, 272–274 Search PubMed , CCDC reference number 796926; (b) S. A. Myz, T. P. Shakhtshneider, N. A. Tumanov, and E. V. Boldyreva, Proceed. X Intern. Conference “Solid state chemistry: nanomaterials, nanotechnologies”, Stavropol, October 17–22, 2010, pp. 351 Search PubMed; (c) S. A. Myz, T. P. Shakhtshneider, N. A. Tumanov, E. V. Boldyreva, Proceed. Research Conference “Nanotechnologies of functional materials”, St. Petersbourg, September, 22–24, 2010, pp. 116–117 Search PubMed; (d) S. A. Myz, N. A. Tumanov, T. P. Shakhtshneider, E. V. Boldyreva, Abstracts Intern. Confer. Chem. Organic Solid State (ICCOSS XX), Bangalore, India, June 26–30, 2011 Search PubMed.
- P. Luger, K. Daneck, W. Engel, G. Trummlitz and K. Wagner, Eur. J. Pharm. Sci., 1996, 4, 175–187 CrossRef CAS.
- M. L. Cheney, D. R. Weyna, N. Shan, M. Hanna, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 4401–4413 CrossRef CAS.
- S. A. Myz, N. A. Tumanov, T. P. Shakhtshneider, E. V. Boldyreva, Russ. Chem. Bull., 2011, in press Search PubMed.
- G. M. Sheldrick, Acta Cryst. A, 2008, 64, 112–122.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS; (b) C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS; (c) F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS; (d) J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Cryst. B, 2004, 60, 627–668 CrossRef; (e) J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC; (f) S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystalExplorer, 2.1., University of Western Australia, Perth, 2007, http://hirshfeldsurfacenet/CrystalExplorer Search PubMed.
- L. Coppi, M. B. Sanmarti and M. C. Clavo, US Patent 6,967,248 B2 USA, 2005.
- T. Mezei, N. Mesterházy, T. Bakó, M. Porcs-Makkay, G. Simig and B. Volk, Org. Process Res. Dev., 2009, 13, 567–572 Search PubMed.
- (a) S. Defazio and R. Cini, J. Chem. Soc., Dalton Trans., 2002, 1888–1897 RSC; (b) S. Defazio and R. Cini, Polyhedron, 2003, 22, 1355–1366 Search PubMed; (c) R. Cini, G. Tamasi, S. Defazio and M. B. Hursthouse, J. Inorg. Biochem., 2007, 101, 1140–1152 CrossRef CAS; (d) G. Tamasi, M. Casolaro, A. Magnani, A. Sega, L. Chiasserini, L. Messori, C. Gabbiani, S. M. Valiahdi, M. A. Jakupec, B. K. Keppler, M. B. Hursthouse and R. Cini, J. Inorg. Biochem., 2010, 104, 799–814 Search PubMed.
- N. Tumanov, M. Dyakonova, N. A. Pankrushina and T. P. Shakhtshneider, 2011, CCDC 832082, deposited in CSD as a private communication.
- F. H. Allen, Acta Crystallogr., 2002, B58, 380–388 CAS.
- S. L. Childs, P. A. Wood, N. Rodríguez-Hornedo, L. S. Reddy and K. I. Hardcastle, Cryst. Growth Des., 2009, 9, 1869–1888 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, 1997, New York, Oxford University Press Search PubMed.
- J. F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee, H. R. Guzman and O. Almarsson, J. Am. Chem. Soc., 2003, 125, 8456–8457 CrossRef CAS.
- (a) W. Ostwald, Z. Phys. Chem., 1879, 22, 289–320 Search PubMed; (b) T. Threlfall, Org. Process Res. Dev., 2003, 7, 1017–1027 Search PubMed.
- (a) A. Alhalaweh and S. P. Velaga, Cryst. Growth Des., 2010, 10, 3302–3305 Search PubMed; (b) N. A. Tumanov, E. V. Boldyreva and N. E. Shikina, Acta Crystallogr., 2010, C66, o279–o283 Search PubMed; (c) A. Alhalaweh, S. George, D. Boström and S. P. Velaga, Cryst. Growth Des., 2010, 10, 4847–4855 Search PubMed; (d) I. A. Tumanov, A. F. Achkasov, E. V. Boldyreva and V. V. Boldyrev, CrystEngComm, 2011, 13, 2213–216 RSC; (e) T. Friščič and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef CAS; (f) D. Braga, S. L. Giaffreda, F. Grepioni, M. R. Chierotti, R. Gobetto, G. Palladino and M. Polito, CrystEngComm, 2007, 9, 879–881 RSC; (g) A. Jayasankar, D. J. Good and N. Rodríguez-Hornedo, Mol. Pharmaceutics, 2007, 4, 360–372 CrossRef CAS; (h) C. Maheshwari, A. Jayasankar, N. A. Khan, G. E. Amidon and N. Rodríguez-Hornedo, CrystEngComm, 2009, 11, 493–500 RSC; (i) F. C. Strobridge, N. Juda and T. Friščić, CrystEngComm, 2010, 12, 2409–2418 RSC; (j) T. Friić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC; (k) D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 1106–1123 CrossRef CAS; (l) K. Guo, G. Sadiq, C. Seaton, R. Davey and Q. Yin, Cryst. Growth Des., 2010, 10, 268–273 CrossRef CAS; (m) E. Gagnière, D. Mangin, F. Puel, A. Rivoire, O. Monnier, E. Garcia and J. P. Klein, J. Cryst. Growth, 2009, 311, 2689–2695 CrossRef CAS; (n) K.-C. Lee and K.-J. Kim, Chem. Eng. Technol., 2011, 34, 619–623 Search PubMed; (o) S. Skovsgaard and A. D. Bond, CrystEngComm, 2009, 11, 444–453 RSC.
- (a) P. J. Wheatley, J. Chem. Soc., 1964, 6017–6022 RSC; (b) Y. Kim, K. Machida, T. Taga and K. Osaki, Chem. Pharm. Bull., 1985, 33, 2641–2647 CAS; (c) C. C. Wilson, New J. Chem., 2002, 26, 1733–1739 RSC; (d) A. Harrison, R. Ibberson, G. Robb, G. Whittaker, C. Wilson and D. Youngson, Faraday Discuss., 2003, 122, 363–393 RSC.
- (a) T. J. Kistenmacher and R. E. Marsh, J. Am. Chem. Soc., 1972, 94, 1340–1345 CrossRef CAS; (b) Z. Galdecki, M. L. Glowka and Z. Gorkiewicz, Farmacja Polska, 1977, 33, 585–587 Search PubMed; (c) P. J. Cox and P. L. Manson, Acta Crystallogr. E, 2003, 59, o986–o988 CrossRef; (d) X. Chen, K. R. Morris, U. J. Griesser, S. R. Byrn and J. G. Stowell, J. Am. Chem. Soc., 2002, 124, 15012–15019 CrossRef CAS.
- A. R. Sheth, S. Bates, F. X. Muller and D. J. W. Grant, Cryst. Growth Des., 2005, 4, 1091–1098 Search PubMed.
- R. E. Marsh, Acta Crystallogr., 1958, 11, 654–63 CrossRef CAS.
- (a) Y. Umeda, T. Fukami, T. Furuishi, T. Suzuki, M. Makimura and K. Tomono, Chem. Pharm. Bull., 2007, 55, 832–836 Search PubMed; (b) Y. Umeda, T. Fukami, T. Furuishi, T. Suzuki, K. Tanjoh and K. Tomono, Drug Dev. Ind. Pharm., 2009, 35, 843–851 Search PubMed; (c) S. L. Childs and K. I. Hardcastle, Cryst. Growth Des., 2007, 7, 1291–1304 CrossRef CAS; (d) E. A. Losev, B. A. Zakharov, T. N. Drebushchak and E. V. Boldyreva, Acta Crystallogr., 2011, C67, o297–o300 Search PubMed; (e) M. A. Mohammad, A. Alhalaweh and S. P. Velaga, Int. J. Pharm., 2011, 407, 63–71 Search PubMed; (f) G. Reck, G. Dietz, G. Laban, W. Gunther, G. Bannier and E. Höhne, Die Pharmazie, 1988, 43, 477 Search PubMed; (g) B. Kojić-Prodić and Ž. Rużić-Toroš, Acta Crystallogr., 1982, B38, 2948–2951 Search PubMed.
- (a) F. Kozjek, L. Golic, P. Zupet, E. Palka, P. Vodopivec and M. Japelj, Acta Pharm. Jugosl, 1985, 35, 275–281 Search PubMed; (b) F. Vrecer, M. Vrbinc and A. Meden, Int. J. Pharm., 2003, 256, 3–15 CrossRef CAS.
- C. B. Aakeroy, S. Forbes and J. Desper, J. Am. Chem. Soc., 2009, 131, 17048–17049 CrossRef CAS.
- D. J. Good and N. Rodriguez-Hornedo, Cryst. Growth Des., 2010, 10, 1028–1032 CrossRef.
- (a) S. J. Bethune, N. Huang, A. Jayasankar and N. Rodriguez-Hornedo, Cryst. Growth Des., 2009, 9, 3976–3988 CrossRef CAS; (b) N. Huang and N. Rodríguez-Hornedo, Cryst. Growth Des., 2010, 10, 2050–2053 CrossRef CAS; (c) A. Jayasankar, L. Roy and N. Rodriguez-Hornedo, J. Pharm. Sci., 2010, 99, 3977–3985 CAS; (d) S. J. Nehm, B. Rodríguez-Spong and N. Rodríguez-Hornedo, Cryst. Growth Des., 2006, 6, 592–600 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 834808 and 834809. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce05902e |
| ‡ Most of the meloxicam co-crystals with a carboxylic acid as a co-former have the 2:1 stoichiometry, which, apparently, reflects the structure-forming unit described further in the text. In case of the co-crystal of meloxicam with glutaric acid (1:1), glutaric acid forms a dimer, which can be taken as ‘another, larger dicarboxylic acid’, and in this case even this structure can be considered as having the same structural motif as the (2:1) series. |
| § The existence of meloxicam polymorphs is mentioned in ref. 15 but it has not been confirmed by the publication of the structural data. In the original source (ref. 19) the existence of five ‘forms’, not five ‘polymorphs’ was claimed. For example, ‘Form I’ in ref. 19 corresponds to the known polymorph of meloxicam,14 but ‘Form IV’ to meloxicam hydrate. The crystal structures and even the chemical composition of the other three ‘Forms’ in ref. 19 were not described. The X-ray powder diffraction patterns reported in ref. 19 for ‘Form II’ and ‘Form V’ correspond to multiphase samples. |
| ¶ Compare this structure-forming fragment with trimers in the co-crystals of a triazole drug with 1,2-dicarboxylic acids.26 |
| || The interpretation of the fine details of the dissolution kinetics, as well as of the existing differences in the dissolution of meloxicam co-crystals with various carboxylic acids (not related to the presence/absence of dimers) requires a detailed analysis, which is beyond scope of the present paper and will be reported elsewhere. |
| This journal is © The Royal Society of Chemistry 2012 |