DOI:
10.1039/C1CE05605K
(Paper)
CrystEngComm, 2012,
14, 314-322
Three novel organic-inorganic hybrid materials based on decaoxovanadates obtained from a new liquid phase reaction†
Received
21st May 2011
, Accepted 3rd October 2011
First published on 9th November 2011
Abstract
Three organic-inorganic hybrid materials based on decavanadates: H4V10O28·(HMTA-CH2OH)2·6H2O (1), H4V10O28·(HMTA-CH2OH)2·4H2O (2) and H2V10O28·(HMTA)2·[Co(H2O)6]2·6H2O (3) (HMTA = hexamethylenetetramine) have been crystallized from a new liquid phase reaction. These complexes display 3D supramolecular networks through various hydrogen bonds and generate typical binodal topologies. Complex 1 was further used as a functional precursor to prepare V, N-containing mesoporous carbon foams (V, N-MCFs), which exhibit highly ordered mesostructures with specific surface areas of 653 m2 g−1 and uniform pore sizes of 2.9 nm. Cyclic voltammetry and charge-discharge tests indicate that V, N-MCFs possess much higher capacitance in comparison with pure mesoporous carbon foams (MCFs).
Introduction
Exploratory synthesis and research on organic–inorganic hybrid materials has been an attractive field due to their potential applications as functional materials, as well as their intriguing variety of architectures and topologies.1 Polyoxovanadates are one kind of significant metal-oxo clusters with nanosizes, abundant networks, interesting electronic and structural properties which have considerable importance in the area of catalysis, biology, geochemistry and material sciences,2 and they have been employed as inorganic building blocks for constructing supramolecular arrays with various organic ligands recently.3 Among the polyoxovanadates, decavanadates are one of the most common polyanions in the presence of a number of organic bases. Experimental studies have shown that whenever the pH of the initial solution is in the range of 4 to 6, decavanadates containing salts are predominantly isolated4 whereas the decavanadate polyanions are fully oxidized and frequently protonated, and the available nucleophilic surface of oxygen atoms on decavanadates interacts with organic ligands to form multiple hydrogen bonds for constructing a high-dimensional supramolecular network.
Herein, we report the synthesis and structural characterization of three organic-inorganic hybrid materials based on decavanadates: H4V10O28·(HMTA-CH2OH)2·6H2O (1), H4V10O28·(HMTA-CH2OH)2·4H2O (2), H2V10O28·(HMTA)2·[Co(H2O)6]2·6H2O (3) (HMTA = hexamethylenetetramine). All of them were obtained from a novel liquid phase reaction (Scheme 1).
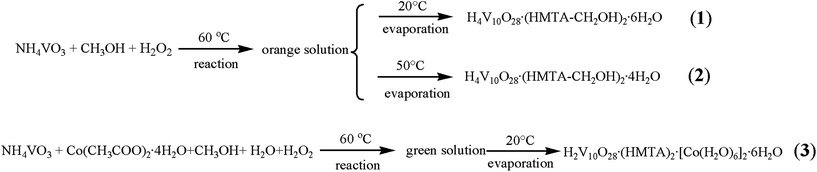 |
| | Scheme 1 Syntheses of 1–3. | |
Porous carbon materials, which possess excellent chemical and physical stability, are very interesting materials for sensors, membranes, separation, catalyst supports, energy conversion and storage.5 Especially, mesoporous carbon foams (MCFs) attract broad attention as supercapacitor electrode materials due to their high specific surface area, abundant mesoporous structure and appropriately porous size for quick mass transfer and ion diffusion.6 However, due to their low electrical conductivity and relatively inert surface, the specific capacitance of MCFs is still limited.7 To improve the electrochemical behaviors, various compounds were introduced into the MCFs.8 For instance, it was reported that the incorporation of vanadium oxides or vanadate salts into MCFs could enhance their electrochemical properties,8h–8j while introducing compounds containing nitrogen8k–8n into MCFs could increase their graphitization degree and consequent conductivity. Complex 1 is easy to prepare and can be dissolved in water, therefore, we employed complex 1 as a functional precursor to prepare V, N-containing mesoporous carbon foams (V, N-MCFs) through an evaporation induced self-assembly approach, and evaluated their electrochemical behaviors through cyclic voltammetry and galvanostatic charge-discharge analysis.
Experimental
Materials and measurements
All the starting reagents were purchased commercially and used without further purification. Elemental analyses were performed on a Perkin-Elmer 2400II elemental analyzer. IR spectra were measured in KBr pellets on a Nicolet 5DX FT-IR spectrometer. Transmission electron microscopy (TEM) images were obtained by using a JEOL JEM-1230. Nitrogen sorption analyses were performed on Micromeritics Tristar 3000, with samples being heated to 200 °C under vacuum (10−5 Torr) for 2 h. Nitrogen adsorption data were taken at the liquid nitrogen temperature using a Micromeritics Tristar 3000 analyzer to calculate the specific surface area by the Brunauer-Emmett-Teller (BET) model. Thermal gravimetric analyses (TGA) were carried out using a Netzsch STA 449C thermoanalysis instrument in the temperature range 30–800 °C. Powder X-ray diffraction (XRD) experiments were taken on a Bruker D8 Advance diffractometer with Cu-Kα 1 (λ = 1.54056 Å) radiation. Raman spectra were obtained by using a Renishaw Invia. system, under λexc = 514 nm laser excitation.
Synthesis of H4V10O28·(HMTA-CH2OH)2·6H2O (1) and H4V10O28·(HMTA-CH2OH)2·4H2O (2)
0.466 g NH4VO3 (4.00 mmol), 40 ml of CH3OH (methanol) and 15 ml of 30% H2O2 were mixed and stirred at 60 °C for 4 h, the resulting orange solution (pH between 3.5 and 4.5) was kept at 20 °C. Orange block crystals of 1 suitable for X-ray crystallographic study were crystallized via slow evaporation in 2 weeks. Yield: 86% based on V. IR (KBr pellets, ν/cm−1): 3428(s), 3209(s), 3001(m), 1631(m), 1407(m), 1385(m), 1261(m), 1230(w), 1106(m), 1072(m), 1037(m), 999(m), 833(s), 744(s), 663(m), 594(m), 528(m), 501(m) and 447(s). Anal. Calcd. for C14H46N8O36V10: C, 11.91%; H, 3.28%; N, 7.93. Found: C, 11.85%; H, 3.24%; N, 8.01. If the resulting orange solution was evaporated at 50 °C, brown block crystals of 2 could be obtained in 2 days. Yield: 68% based on V. IR (KBr pellets, ν/cm−1): 3425(s), 2923(s), 2854(m), 1639(s), 1403(m), 1303(w), 1257(m), 1218(w), 1022(m), 983(m), 960(s), 744(m), 586(m) and 509(m). Anal. Calcd. for C14H42N8O34V10: C, 12.22%; H, 3.08%; N, 8.14. Found: C, 12.15%; H, 3.04%; N, 8.21.
Synthesis of H2V10O28·(HMTA)2·[Co(H2O)6]2·6H2O (3)
0.466 g NH4VO3 (4.00 mmol), 0.125 g Co(CH3COO)2·4H2O (0.50 mmol), 30 ml CH3OH, 15 ml H2O2 and 10 ml of distilled water were mixed and stirred at 60 °C for 4 h, the resulting green solution (pH between 3.5 and 4.5) was kept at 20 °C. Yellow block crystals suitable for X-ray crystallographic study were separated via slow evaporation in 2 weeks. Yield: 16% based on V. IR (KBr pellets, ν/cm−1): 3428(s), 3228(m), 2957(m), 1601(m), 1487(m), 1344(m), 1305(w), 1272(w), 1207(m), 1126(m), 1010(s), 985(m), 959(s), 855(m), 732(w), 689(w) and 655(s). Anal. Calcd. for C12H62Co2N8O46V10: C, 8.57%; H, 3.72%; N, 6.66. Found: C, 8.50%; H, 3.63%; N, 6.71.
Preparation of V, N-MCFs and MCFs
V, N-MCFs were prepared by using resol as an organic precursor, complex 1 as a functional precursor and triblock copolymers Pluronic P123 (Mw = 5800, EO20–PO70–EO20) as a template. The resol was prepared according to the procedure described in the literature.5b Typically, 1.00g of P123 and 0.03g complex 1 was dissolved in 25 g of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio of ethanol and water mixture. Then 5.00 g of resol precursors in ethanol solution containing 0.61 g of phenol and 0.39 g of formaldehyde was added into above mixture. A homogeneous solution was obtained after stirring for 20 min. The solution was coated as a thin polymer membrane on the surface of a dish and the membrane was kept at 25 °C for 8 h. Thermal polymerization of the membranes was carried out at 100 °C for 12 h. The thin membrane was crushed into powders and carbonized at 800 °C in a purified nitrogen flow with a heat rate of 1 °C min−1 to decompose the surfactant template and to obtain V, N-MCFs. As a comparison, we synthesized MCFs by a similar approach without a functional precursor.
X-ray single crystal data were collected on a Bruker APEX II diffractometer equipped with a graphite–monochromatized Mo-Kα radiation (λ = 0.71073 Å) at 296(2) K. Intensity data were corrected by Lorentz-polarization factors and empirical absorption. The structures were solved by direct methods and expanded with difference Fourier techniques. All non-hydrogen atoms were refined anisotropically. Except for the hydrogen atoms on oxygen atoms which were located from the difference Fourier maps, the other hydrogen atoms were generated geometrically. All calculations were performed using SHELXS-97 and SHELXL-97.9 Further details for structural analyses are summarized in Table 1. CCDC-685560 (1), 746415 (2) and 745997 (3) contain the crystallographic data in CIF format.
:1 weight ratio of ethanol and water mixture. Then 5.00 g of resol precursors in ethanol solution containing 0.61 g of phenol and 0.39 g of formaldehyde was added into above mixture. A homogeneous solution was obtained after stirring for 20 min. The solution was coated as a thin polymer membrane on the surface of a dish and the membrane was kept at 25 °C for 8 h. Thermal polymerization of the membranes was carried out at 100 °C for 12 h. The thin membrane was crushed into powders and carbonized at 800 °C in a purified nitrogen flow with a heat rate of 1 °C min−1 to decompose the surfactant template and to obtain V, N-MCFs. As a comparison, we synthesized MCFs by a similar approach without a functional precursor.
X-ray single crystal data were collected on a Bruker APEX II diffractometer equipped with a graphite–monochromatized Mo-Kα radiation (λ = 0.71073 Å) at 296(2) K. Intensity data were corrected by Lorentz-polarization factors and empirical absorption. The structures were solved by direct methods and expanded with difference Fourier techniques. All non-hydrogen atoms were refined anisotropically. Except for the hydrogen atoms on oxygen atoms which were located from the difference Fourier maps, the other hydrogen atoms were generated geometrically. All calculations were performed using SHELXS-97 and SHELXL-97.9 Further details for structural analyses are summarized in Table 1. CCDC-685560 (1), 746415 (2) and 745997 (3) contain the crystallographic data in CIF format.
| Complex |
1
|
2
|
3
|
| Empirical formula |
C14H46N8O36V10 |
C14H42N8O34V10 |
C12H62Co2N8O46V10 |
| Formula weight |
1411.99 |
1375.96 |
1681.96 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
C2/c |
|
a/Å |
9.4462(4) |
12.0418(3) |
22.0808(15) |
|
b/Å |
10.6899(4) |
15.7976(3) |
9.7061(5) |
|
c/Å |
10.9170(4) |
11.0989(3) |
24.3647(15) |
|
α (°) |
103.250(2) |
90 |
90 |
|
β (°) |
98.612(2) |
107.98(0) |
109.045(8) |
|
γ (°) |
102.932(2) |
90 |
90 |
|
V/Å3 |
1022.07(7) |
2008.25(7) |
4936.0(5) |
|
Z
|
1 |
2 |
4 |
|
D
c
|
2.294 |
2.275 |
2.263 |
|
μ/mm−1 |
2.301 |
2.335 |
2.581 |
|
F(000) |
704 |
1368 |
3368 |
|
θ
min, θmax (°) |
1.96, 27.52 |
1.78, 27.72 |
2.59, 27.57 |
| Reflections collected |
17412 |
31667 |
36884 |
| Unique reflections (Rint) |
4686 (0.0268) |
4693 (0.0500) |
5695 (0.0833) |
| Observed reflections |
|
|
|
| (I > 2σ(I)) |
3889 |
3797 |
4079 |
| Parameters refined |
334 |
319 |
416 |
|
S on F2 |
1.013 |
1.001 |
1.010 |
|
R, wR (I > 2σ(I)) |
0.0295, 0.0792 |
0.0352, 0.0932 |
0.0490, 0.1322 |
|
R, wR (all data) |
0.0387, 0.0859 |
0.0469, 0.1004 |
0.0750, 0.1499 |
Electrochemical evaluation
The electrochemical measurements were done in a three-electrode experimental set-up. A saturated calomel electrode (SCE) was used as the reference electrode, and nickel foil as the counter-electrode. The working electrode was prepared as follows: V, N-MCFs or MCFs powders were mixed with poly(tetrafluoroethylene) and graphite with a mass ratio of 8.5:1:0.5. The mixture was pressed between two pieces of nickel foam under 30 MPa. Thereafter, the electrode was dried overnight at 100 °C. Cyclic voltammograms and galvanostatic charge-discharge behavior were tested in the electrolyte of 1.0 mol L−1Na2SO4 aqueous solution using a CHI 660D electrochemical workstation, and the potential window was chosen in the range of 0–0.6 V versusSCE. The specific capacitance C (F/g) is calculated from the gravimetric discharge process according to the equation: C = (i × Δtd)/(m × ΔV) where i is the constant current (A), m is the mass of the activated substance (g), Δtd is the discharging time (s), and ΔV is the potential change (V). The potential range we used to estimate was 0.2 to 0.4 V.
Results and discussion
Synthesis
Our synthetic strategy for complexes 1, 2 and 3 are depicted in Scheme 1. These three hybrid compounds were obtained from similar liquid phase reactions, in which methanol and NH4+ should be oxidized to [HMTA-CH2OH]+ or HMTA by H2O2 with the existence of vanadate. There are more than four hundred organometallic polymers containing HMTA groups in the Cambridge Structural Database,10 most of the HMTA groups in these structures came from original organic ligands, and three of which are hybrid materials based on polyoxovanadates,.4a,4j It is notable that the [HMTA-CH2OH]+ or HMTA groups in complexes 1–3 came from in situ oxidization of methanol and NH4+. Besides, if sufficient NH3·H2O was added into the resulting solution subsequently (Scheme 2), rhombus colorless crystals of HMTA can be further separated.11HMTA is an important chemical product which is widely used in the textile and plastics industry, organic synthesis as well as the pharmaceutical industry. In general, HMTA can be generated by condensation of formaldehyde and ammonia in alkaline media and will be hydrolyzed in an acid medium.12 However; it is much harder to obtain HMTA from methanol by a one step reaction,13 to our best knowledge, there is no reported case about these unusual reactions which can be carried out smoothly under mild conditions with the advantages of low cost, quick action and simple steps, this new liquid phase reaction may have potential industrial applications.14
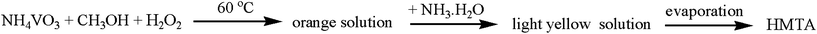 |
| | Scheme 2 Syntheses of HMTA. | |
Crystal structure
H4V10O28·(HMTA-CH2OH)2·6H2O (1).
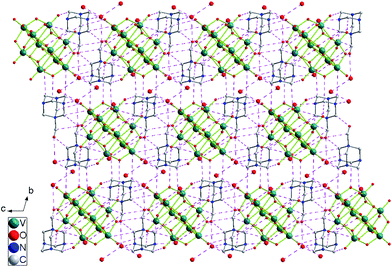
Single-crystal X-ray diffraction analysis reveals that complex 1 crystallizes in the triclinic space groupP. The crystallographically independent unit contains a half protonized [H4O28V10]2− anion, one [HMTA-CH2OH]+ cation as well as three lattice water molecules. As shown in Fig. 1, the centrosymmetric [H4O28V10]2−cluster anion lying about an inversion centre contains ten [VO6] octahedral combined via shared edges and shared corners, and the V–O bond distances fall in the range of 1.599(7)–2.345(3) Å which are comparable to those in [HnV10O28](6−n)− reported in the literature.4 The [HMTA-CH2OH]+ cations, which are counter-ions of the [H4O28V10]2− anions, are located in the gaps with lattice water molecules and provide various (O–H⋯O, N–H⋯O and C–H⋯O) hydrogen bonds (Table 3) to link [H4O28V10]2− anions and generate the novel 3D supramolecular network. (Fig. 2)
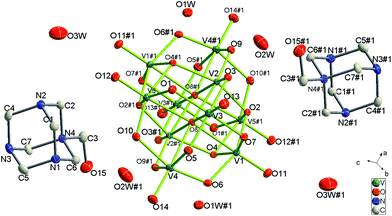 |
| | Fig. 1 Structure of 1 showing the atom labeling, thermal ellipsoids are shown at the 30% probability level. All the hydrogen atoms are omitted for clarity. Symmetry code for the generated atoms: (#1) 1 − x, 1 − y, 1 − z. | |
 |
| | Fig. 2 View down the a axis of the 3-D supramolecular network of 1, showing the multiple hydrogen bonding interactions (broken lines), all H atoms are omitted for clarity. | |
In the topological view, the most interesting structural feature of 1 is the unusual 3D network assembled from [HMTA-CH2OH]+ cations and decavanadate [H4O28V10]2− anions through various hydrogen bonds. As shown in Fig. 3, each [HMTA-CH2OH]+ cation is linked to four [H4O28V10]2− anions and one neighboring [HMTA-CH2OH]+ cation to represent a 5-connected node (Fig. 3a), meanwhile each [H4O28V10]2− anion serves for a 8-connected node by combining eight nearby [HMTA-CH2OH]+ cations (Fig. 3b). In this way, a non-interpenetrating (5,8)-connected network with Schläfli symbol of (32.410.58.64.74)(32.46.52)2 is formed (Fig. 3c). The extended point symbol calculation using TOPOS 4.0[9] gives (3. 3. 4. 4. 4. 4. 4. 4. 52. 52) for the 5-connected node and (3. 3. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 5. 5. 5. 5. 5. 5. 5. 5. 64. 64. 65. 65. 72. 72. 78. 78) for the 8-connected node. This resulting binodal (5,8)-connected topology is known as the rare case of the topological type 5,8T2. To date, only 3 metal organic frameworks15 showing 5,8T2 topology are collected on the TOPOS16 databases, all of them are composed by coordination bonds, complex 1 is the first example of 5,8T2 topology dominated by hydrogen bonds. Besides, it is noteworthy that this network possesses 1D quadrangle channels running parallel to the c axis, which are occupied by the lattice waters.
![(a) 5-connected [HMTA-CH2OH]+ cation linked with four [H4O28V10]2− anions and one neighboring [HMTA-CH2OH]+ cation; (b) 8-connected [H4O28V10]2− anion linked with eight [HMTA-CH2OH]+ cations. (c) Schematic representation of the (5,8)-connected network of 1.](/image/article/2012/CE/c1ce05605k/c1ce05605k-f3.gif) |
| | Fig. 3 (a) 5-connected [HMTA-CH2OH]+ cation linked with four [H4O28V10]2− anions and one neighboring [HMTA-CH2OH]+ cation; (b) 8-connected [H4O28V10]2− anion linked with eight [HMTA-CH2OH]+ cations. (c) Schematic representation of the (5,8)-connected network of 1. | |
H4V10O28·(HMTA-CH2OH)2·4H2O (2).
Complex 2 crystallizes in the monoclinic P21/c space group. As shown in Fig. 4, each crystallographically independent unit contains a half protonized [H4O28V10]2− anion, one [HMTA-CH2OH]+ cation and two lattice water molecules. The [H4O28V10]2− anion also lies about an inversion centre in this structure, and both bond lengths and angles of the centrosymmetric [H4V10O28]2− anion are similar to ones in complex 1. The distance between neighboring two [H4V10O28]2− anions is closer than that of complex 1, therefore there are weak hydrogen bonding interactions (O13–H13⋯O14#6) between these cluster anions. [HMTA-CH2OH]+ cations and lattice water molecules are located in the narrower gaps providing multiple hydrogen bonds and further stabilize the supramolecular structure (Fig. 4, Fig. 5).
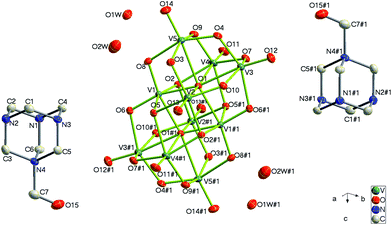 |
| | Fig. 4 Structure of 2 showing the atom labeling, thermal ellipsoids are shown at the 30% probability level. All the hydrogen atoms and lattice water molecules are omitted for clarity. Symmetry code for the generated atoms: (#1) 1 − x, 1 − y, 1 − z. | |
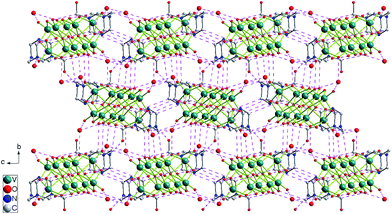 |
| | Fig. 5 View down the a axis of the 3D supramolecular network of 2, showing the multiple hydrogen bonding interactions (broken lines), all H atoms are omitted for clarity. | |
Compared with complex 1, the network of 2 is more complicated. As shown in Fig. 6, each [HMTA-CH2OH]+ cation is linked to four [H4O28V10]2− anions and two neighboring [HMTA-CH2OH]+ cations to represent a 6-connected node; each [H4O28V10]2− anion serves for a 12-connected node by combining eight nearby [HMTA-CH2OH]+ cations and four neighboring [H4O28V10]2− anions through hydrogen bonds. The topology analysis shows that the [HMTA-CH2OH]+ cation and [H4O28V10]2− anion have the Schläfli symbol (36.48.5)2 and (312.424.520.69.7) [extended point symbol (3. 3. 3. 3. 3. 3. 4. 4. 4. 4. 4. 4. 4. 43. 56) and (3. 3. 3. 3. 3. 3. 3. 3. 3. 3. 3. 3. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4. 4(3). 4(3). 5. 5. 5. 5. 5. 5. 5. 5. 52. 52. 52. 52. 52. 52. 52. 52. 52. 52. 54. 54. 64. 64. 68. 68. 68. 69. 69. 69. 69. 74)], respectively, to give rise to a novel 3D (6,12)-connected net (Fig. 6c), which has been deposited in the TOPOS database (rcsr.anu.edu.au) as fly1. In this topological structure, all the water molecules are located at the 1D quadrangle channels running parallel to the c axis.
![(a) 6-connected [HMTA-CH2OH]+ cation linked with four [H4O28V10]2− anions and two neighboring [HMTA-CH2OH]+ cations; (b) 12-connected [H4O28V10]2− anion linked with eight [HMTA-CH2OH]+ cations and four neighboring [H4O28V10]2− anions; (c) Schematic representation of the (6, 12)-connected network of 2.](/image/article/2012/CE/c1ce05605k/c1ce05605k-f6.gif) |
| | Fig. 6 (a) 6-connected [HMTA-CH2OH]+ cation linked with four [H4O28V10]2− anions and two neighboring [HMTA-CH2OH]+ cations; (b) 12-connected [H4O28V10]2− anion linked with eight [HMTA-CH2OH]+ cations and four neighboring [H4O28V10]2− anions; (c) Schematic representation of the (6, 12)-connected network of 2. | |
To the best of our knowledge, the construction of binodal nets with high coordination numbers is still a challenging issue in coordination chemistry.17 The RCSR database contains only 17 high-connected binodal nets that are not edge transitive and have a node degree larger than eight. In 2009, the analysis by Blatov and Proserpio revealed that only 18 binodal nets (0.39%) containing two 6–12-coordinated nodes are found (one alb and 17 nia nets) among the 2131 three-dimensional metal–organic polymeric frameworks and 2473 hydrogen-bonded organic and metal–organic frameworks.17a Thus, the discovery of this new topology fly1 is fundamental in the crystal engineering of supramolecular networks.
H2V10O28·(HMTA)2·[Co(H2O)6]2·6H2O (3).
Complex 3 crystallizes in the monoclinic C2/c space group. As shown in Fig. 7, each crystallographically independent unit contains a half protonized [H2O28V10]4− anion, one [Co(H2O)6]2+ cation, one HMTA molecule and three lattice water molecules. In this structure, the [H2O28V10]4− anion lies about a twofold axis. The Co–O bond distances fall in the range of 2.049(4)–2.124(3) Å and the values of cis –O–Co–O angles are between 88.10(14) and 92.68(18)°. As shown in Fig. 8, HMTA and water molecules connect [H2O28V10]4− anions and [Co(H2O)6]2+ cations and further generate the supramolecular network through multiple hydrogen bonds.
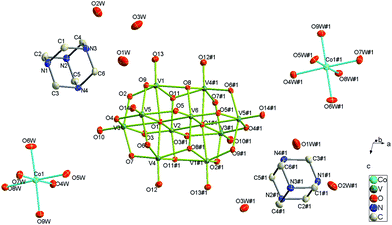 |
| | Fig. 7 Structure of 4 showing the atom labeling, thermal ellipsoids are shown at the 30% probability level. All the hydrogen atoms and lattice water molecules are omitted for clarity. Symmetry code for the generated atoms: (#1) 1 − x, y, 0.5 − z. | |
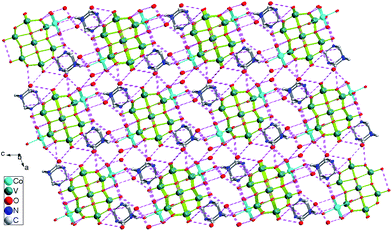 |
| | Fig. 8 View down the b axis of the 3-D supramolecular network of 4, showing the multiple hydrogen bonding interactions (broken lines), all H atoms are omitted for clarity. | |
As shown in Fig. 9, each HMTA molecule is linked to four [H2O28V10]4− anions to represent a 4-connected node; each [H2O28V10]4− anion serves for a 8-connected node by combining eight nearby HMTA molecules through hydrogen bonds; thus, a well-known binodal (4, 8)-connected topology of fluorite (flu) network with Schläfli symbol of (412.612.84)(46)2 is formed. Additionally, it is worth mentioning that this network possesses three types of 1D quadrangle channels running parallel to the b axis, the smallest channels are empty, the medium-sized channels are filled with water molecules, while the largest ones are occupied by the hydrated [Co(H2O)6]2+ cations and lattice water molecules (Fig. 9c)
![(a) 4-connected HMTA molecule linked with four [H4O28V10]2− anions; (b) 8-connected [H4O28V10]2− anion linked with eight HMTA molecules; (c) Schematic representation of the (4, 8)-connected network of 3.](/image/article/2012/CE/c1ce05605k/c1ce05605k-f9.gif) |
| | Fig. 9 (a) 4-connected HMTA molecule linked with four [H4O28V10]2− anions; (b) 8-connected [H4O28V10]2− anion linked with eight HMTA molecules; (c) Schematic representation of the (4, 8)-connected network of 3. | |
Structure of mesoporous carbon foam
Fig. 10 shows the nitrogen adsorption and desorption isotherms and pore size distribution curves of V, N-MCFs and MCFs. The isotherms of V, N-MCFs show representative type IV curves18 which exhibit a sharp capillary condensation step at relative pressures of 0.4–0.6, corresponding to the narrow pore size distribution. The V, N-MCFs possess the Brunauer-Emmett-Teller (BET) surface area of 653 m2 g−1. The pore size distribution of V, N-MCFs, which is calculated from the desorption branch of the isotherm using the Barrett-Joyner-Halenda method, exhibits single pores with the most probable pore size of 2.9 nm. For comparison, MCFs possess the BET area of 725 m2 g−1 and the most probable pore size of 3.2 nm. These results reveal that V, N-MCFs have mesoporous structure and possess uniform pore size, and the functionalization has no obvious influence on the framework of V, N-MCFs. Fig. 11 shows the transmission electron microscopy (TEM) image of V, N-MCFs. It indicates that V, N-MCFs have a highly ordered mesostructure with uniform pores and the individual pore size is around 3 nm, which matches the results of nitrogen adsorption and desorption analyses. As shown in Fig. S1†, the TG curve of complex 1 under air atmosphere indicates that complex 1 was decomposed and reduced up to 50 °C, and the final residuals may be V2O5, the observed total weight loss of 35.19% is close to the calculated one of 35.60%. As shown in Fig. S2†, the TG curve of V, N-MCFs under air atmosphere indicates that the amount of vanadium in the V, N-MCFs was around 2.4 wt.%. The mass ratio of C (83 wt%) H (2.8 wt%) and N (1.1 wt%) in V, N-MCFs were detected by CHN elemental analysis. As shown in Fig. S3†, the wide-angle XRD patterns of V, N-MCFs and MCFs are similar. Two broad diffraction peaks located around 24° and 44° are observed in both V, N-MCFs and MCFs which can be assigned to the (002) and (100) lattice planes of the graphite structure. There is no other significant peak in the case of V, N-MCFs, which is due to the limited content of impurities. The Raman spectra of V, N-MCFs and MCFs were also analyzed. As shown in Fig. S4†, both of the two spectra show a distinct pair of broad bands near 1580 cm−1 (G band) and 1337 cm−1 (D band). The G band and D band are assigned to the hexagonal carbon plane and crystal defects or imperfections, respectively. The ratio of the relative intensity of these two bands (ID/IG) is proportional to the number of defect sites in the graphite carbon. The lower the ratio is, the higher the graphitization is. It can be calculated that the ID/IG ratio declines from 1.06 (MCFs) to 0.93 (V, N-MCFs), which shows that the graphitization degree of V, N-MCFs is increased in comparison with MCFs. These results indicate that the electrical conductivity of V, N-MCFs was consequently improved.8h
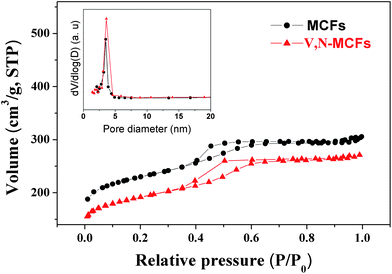 |
| | Fig. 10
Nitrogen adsorption and desorption isotherms and the pore size distributions (insert) of V, N-MCFs and MCFs. | |
 |
| | Fig. 11
TEM photograph of V, N-MCFs. | |
Electrochemical properties
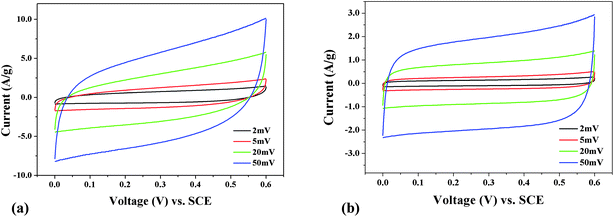
The electrochemical performances of the prepared V, N-MCFs and MCFs were investigated by cyclic voltammetry and galvanostatic charge-discharge tests. Fig. 12 shows the cyclic voltammetry (CV) curves of V, N-MCFs and MCFs electrodes, and the potential window was chosen in the range of 0–0.6 V versusSCE. It is well-known that the specific capacitance is an important parameter to evaluate the performance of electrochemical supercapacitors and the capacitance of material is in proportion to the areas of its CV curves, it can be observed that the capacitance of the V, N-MCFs (Fig. 12a) is much larger than that of MCFs (Fig. 12b). The CV curves of V, N-MCFs were characterized with rectangle-like shape at sweep rates of 2 and 5 mV s−1, which indicates an ideal capacitance behavior;19 however, they veered away much from rectangle-shape at scanning rates of 20 mV s−1 and 50mV s−1, suggesting resistance-like electrochemical behavior.20
The galvanostatic charge-discharge behaviors reflect substantial electrical energy storage; based on the results of charge-discharge cycling, the specific discharge capacitance of a single electrode in the capacitors can be calculated. As shown in Fig. 13, both V, N-MCFs and MCFs electrodes present linear galvanostatic charge-discharge curves at the loading current density of 200 mA g−1. The calculated specific capacitance of the V, N-MCFs reaches 113 F g−1, much higher than that of the MCFs (72 F g−1). These results proved that the specific capacitance of V, N-MCFs was significantly improved compared with MCFs.
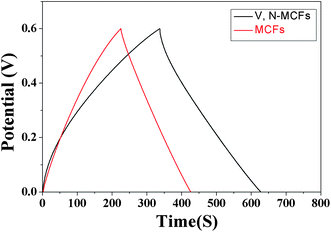 |
| | Fig. 13 Galvanostatic charge-discharge curves of the V, N-MCFs and MCFs electrodes at the loading current density of 200 mA g−1. | |
Conclusion
In summary, a novel liquid phase reaction resulted in the formation of three organic-inorganic hybrid materials based on decavanadates: H4V10O28·(HMTA-CH2OH)2·6H2O (1), H4V10O28·(HMTA-CH2OH)2·4H2O (2), H2V10O28·(HMTA)2·[Co(H2O)6]2·6H2O (3). Complexes 1, 2 and 3 display 3D supramolecular networks with three different binodal topologies through various hydrogen bonds. Complex 1 was further used as a functional precursor to obtain V, N-MCFs, which exhibit highly ordered mesostructure with specific surface areas of 653 m2 g−1 and uniform pore sizes of 2.9 nm. Compared with MCFs, V, N-MCFs possess much higher specific discharge capacitance. It can be expected that V, N-MCFs have a promising prospect for application in supercapacitors.
Acknowledgements
The project was supported by the Science and Technology Department of Zhejiang Province for financial support of the project (2007C21174), the National Natural Science Foundation of China (20973127) and Shanghai Nanotechnology Promotion Center (0952nm00800). The authors are also grateful to Prof. V. A. Blatov for the help about topological analysis.
References
-
(a) M. O'Keeffe and S. T. Hyde, Zeolites, 1997, 19, 370 CrossRef CAS;
(b) M. O'Keeffe, Nature, 1999, 400, 617 CrossRef CAS;
(c) B. Moulton and M. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS;
(d) N. W. Ockwig, O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS;
(e) H. J. Cowley, M. S. Holt, R. L. Melen, J. M. Rawson and D. S. Wright, Chem. Commun., 2011, 47, 2682 RSC;
(f) Y. K. Lv, C. H. Zhan and Y. L. Feng, CrystEngComm, 2010, 12, 3052 RSC;
(g) Q. A. Huang, J. C. Yu, J. K. Gao, X. T. Rao, X. L. Yang, Y. J. Cui, C. D. Wu, Z. J. Zhang, S. C. Xiang, B. L. Chen and G. D. Qian, Cryst. Growth Des., 2010, 10, 5291 CrossRef CAS;
(h) S. Eslava, F. Hengesbach, M. McPartlin and D. S. Wright, Chem. Commun., 2010, 46, 4701 RSC;
(i) Y. K. Lv, Y. L. Feng, J. W. Liu and Z. G. Jiang, J. Solid State Chem., 2011, 184, 1339 CrossRef CAS;
(j) Y. K. Lv, J. Chen, X. J. Wang and Y. L. Feng, Chinese J. Struc. Chem., 2010, 29, 1483 CAS.
-
(a) N. Bosnjakovic-Pavlovic, A. Spasojevic-de Bire, I. Tomaz, N. Bouhmaida, F. Avecilla, U. B. Mioc, J. C. Pessoa and N. E. Ghermani, Inorg. Chem., 2009, 48, 9742 CrossRef CAS;
(b) L. C. Cantley, L. Josephson, R. Warner, M. Yanagisawa, C. Lechene and G. Guidotti, J. Biol. Chem., 1977, 252, 7421 CAS;
(c) F. Carn, M. Djabourov, T. Coradin, J. Livage and N. Steunou, J. Phys. Chem. B, 2008, 112, 12596 CrossRef CAS;
(d) B. X. Dong, J. Peng, C. J. Gomez-Garcia, S. Benmansour, H. Q. Jia and N. H. Hu, Inorg. Chem., 2007, 46, 5933 CrossRef CAS;
(e) C. E. Heyliger, A. G. Tahiliani and J. H. McNeill, Science, 1985, 227, 1474 CAS;
(f) M. I. Khan, E. Yohannes and R. J. Doedens, Angew. Chem., Int. Ed., 1999, 38, 1292 CrossRef CAS;
(g) C. A. Smith and I. Rayment, Biochemistry, 1996, 35, 5404 CrossRef CAS.
-
(a) L. A. Truflandier, F. Boucher, C. Payen, R. Hajja, Y. Millot, C. Bonhomme and N. Steunou, J. Am. Chem. Soc., 2010, 132, 4653 CrossRef CAS;
(b) N. Steens, A. M. Ramadan, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2010, 39, 585 RSC;
(c) N. Suaud, Y. Masaro, E. Coronado, J. M. Clemente-Juan and N. Guihery, Eur. J. Inorg. Chem., 2009, 5109 CrossRef CAS;
(d) M. I. Khan, S. Ayesh, R. J. Doedens, M. H. Yu and C. J. O'Connor, Chem. Commun., 2005, 4658 RSC;
(e) M. I. Khan, S. Tabussum, R. J. Doedens, V. O. Golub and C. J. O'Connor, Inorg. Chem. Commun., 2004, 7, 54 CrossRef CAS;
(f) T. Kurata, Y. Hayashi and K. Isobe, Chem. Lett., 2010, 39, 708 CrossRef CAS.
-
(a) T. Duraisamy, N. Ojha, A. Ramanan and J. J. Vittal, Chem. Mater., 1999, 11, 2339 CrossRef CAS;
(b) Z. H. Yi, X. Y. Yu, W. J. Xia, L. Y. Zhao, C. Yang, Q. Chen, X. L. Wang, X. Z. Xu and X. Zhang, CrystEngComm, 2010, 12, 242 RSC;
(c) H. Choi, Y. U. Kwon and O. H. Han, Chem. Mater., 1999, 11, 1641 CrossRef CAS;
(d) M. Del Arco, S. Gutierrez, C. Martin, V. Rives and J. Rocha, J. Solid State Chem., 2000, 151, 272 CrossRef CAS;
(e) T. Douglas and M. Young, Nature, 1998, 393, 152 CrossRef CAS;
(f) P. Parhi, S. Upreti and A. Ramanan, Cryst. Growth Des., 2010, 10, 5078 CrossRef CAS;
(g) V. Rives and S. Kannan, J. Mater. Chem., 2000, 10, 489 RSC;
(h) L. M. Zheng, Y. S. Wang, X. Q. Wang, J. D. Korp and A. J. Jacobson, Inorg. Chem., 2001, 40, 1380 CrossRef CAS;
(i) Y. K. Lu and Y. L. Feng, Chin. J. Chem., 2010, 28, 2404 CrossRef CAS;
(j) T. Duraisamy, A. Ramanan and J. J. Vittal, Cryst. Eng., 2000, 3, 237 CrossRef CAS.
-
(a) M. X. Liu, L. H. Gan, C. Tian, J. C. Zhu, Z. J. Xu, Z. X. Hao and L. W. Chen, Carbon, 2007, 45, 3045 CrossRef CAS;
(b) Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053 CrossRef CAS;
(c) A. I. Cooper, Adv. Mater., 2003, 15, 1049 CrossRef CAS;
(d) K. P. Gierszal and M. Jaroniec, J. Am. Chem. Soc., 2006, 128, 10026 CrossRef CAS;
(e) S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 414, 470 CrossRef CAS;
(f) Y. J. Kim, B. J. Lee, H. Suezaki, T. Chino, Y. Abe, T. Yanagiura, K. C. Park and M. Endo, Carbon, 2006, 44, 1592 CrossRef CAS;
(g) J. Lee, J. Kim and T. Hyeon, Chem. Commun., 2003, 1138 RSC;
(h) J. Lee, J. Kim, Y. Lee, S. Yoon, S. M. Oh and T. Hyeon, Chem. Mater., 2004, 16, 3323 CrossRef CAS;
(i) J. Lee, K. Sohn and T. Hyeon, Chem. Commun., 2002, 2674 RSC;
(j) L. Radhakrishnan, J. Reboul, S. Furukawa, P. Srinivasu, S. Kitagawa and Y. Yamauchi, Chem. Mater., 2011, 23, 1225 CrossRef CAS;
(k) M. Hu, J. Reboul, S. Furukawa, L. Radhakrishnan, Y. Zhang, P. Srinivasu, Hideo Iwai, H. Wang, Y. Nemoto, N. Suzuki, S. Kitagawa and Y. Yamauchi, Chem. Commun., 2011, 47, 8124 RSC;
(l) B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456 CrossRef CAS.
-
(a) S. W. Hwang and S. H. Hyun, J. Non-Cryst. Solids, 2004, 347, 238 CrossRef CAS;
(b) B. Xu, F. Wu, R. Chen, G. Cao, S. Chen and Y. Yang, J. Power Sources, 2010, 195, 2118 CrossRef CAS.
-
(a) M. Inagakia, H. Konnoa and O. Tanaike, J. Power Sources, 2010, 195, 7880 CrossRef;
(b) N. D. Kim, W. Kim, J. B. Joo, S. Oh, P. Kim, Y. Kim and J. Yi, J. Power Sources, 2008, 180, 671 CrossRef CAS.
-
(a) A. Vinu, K. Ariga, T. Mori, T. Nakanishi, S. Hishita, D. Golberg and Y. Bando, Adv. Mater., 2005, 17, 1648 CrossRef CAS;
(b) Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 1553 CrossRef CAS;
(c) A. B. Chen, W. P. Zhang, Y. Liu, X. W. Han and X. H. Bao, Chin. Chem. Lett., 2007, 18, 1017 CrossRef CAS;
(d) S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169 CrossRef CAS;
(e) H. F. Li, R. D. Wang and R. Cao, Microporous Mesoporous Mater., 2008, 111, 32 CrossRef CAS;
(f) Y. Wang, L. Cheng, F. Li, H. Xiong and Y. Xia, Chem. Mater., 2007, 19, 2095 CrossRef CAS;
(g) Y. Doi, A. Takai, S. Makino, L. Radhakrishnan, N. Suzuki, W. Sugimoto, Y. Yamauchi and K. Kuroda, Chem. Lett., 2010, 39, 777 CrossRef CAS;
(h) Y. K. Lv, M. X. Liu, L. H. Gan, Y. J. Cao, L. H. Chen, W. Xiong, Z. J. Xu, Z. X. Hao, H. L. Liu and L. W. Chen, Chem. Lett., 2011, 40, 236 CrossRef CAS;
(i) L. Yu, C. Zhao, X. Long and W. Chen, Microporous Mesoporous Mater., 2009, 126, 58 CrossRef CAS;
(j) L. Hu, L. Yu, C. Zhao, X. Long and W. Chen, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2010, 25, 574 CrossRef CAS;
(k) C. M. Feng, H. X. Li and Y. Wan, J. Nanosci. Nanotechnol., 2009, 9, 1558 CrossRef CAS;
(l) N. D. Kim, W. Kim, J. B. Joo, S. Oh, P. Kim, Y. Kim and J. Yi, J. Power Sources, 2008, 180, 671 CrossRef CAS;
(m) T. Kwon, H. Nishihara, H. Itoi, Q. H. Yang and T. Kyotani, Langmuir, 2009, 25, 11961 CrossRef CAS;
(n) Y. Zhang, C. G. Liu, B. Wen, X. Y. Song and T. J. Li, Mater. Lett., 2011, 65, 49 CrossRef CAS.
-
(a)
G. M. Sheldrick, SHELXL-97, Program for X-Ray Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b)
G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
-
http://www.ccdc.cam.ac.uk/products/csd/
.
- Faming Zhuanli Shenqing Gongkai Shuomingshu, 101735226.
-
(a) DE10103770-A1; SK200200080-A3; NL1019840-C2; DE10103770-B4; IT1332939-B;
(b) DE4121382-C1; DE4121382-C;
(c) D. M. Musher and D. P. Griffith, Antimicrob. Agents Ch., 1974, 6, 708 CAS;
(d) J. G. Strom and H. W. Jun, J. Pharm. Sci., 1980, 69, 1261 CrossRef CAS;
(e) J. G. Strom and H. W. Jun, Biopharm. Drug Dispos., 1993, 14, 61 CrossRef CAS.
-
(a) D. Milstein and J. Mol, Catal., 1986, 36, 387 CAS;
(b) G. X. Zhao, M. Q. Li and M. J. Sun, J. Shanxi Univ. (Nat. Sci. Ed.), 1990, 13, 377 CAS;
(c) RU2198887-C1;
(d) A. Adach, M. Daszkiewicz, B. Barszcz, M. Cieslak-Golonka and G. Maciejewska, Inorg. Chem. Commun., 2010, 13, 361 CrossRef CAS.
-
(a)
D. D. Ridley, Information Retrieval: SciFinder and SciFinder Scholar, Wiley, Chichester, 2002, ISSN 0479-84351-9 Search PubMed;
(b)
http://www.cas.org/SCIFINDER/SCHOLAR/
.
-
(a) S. Muniappan, S. Lipstman, S. George and I. Goldberg, Inorg. Chem., 2007, 46, 5544 CrossRef CAS;
(b) L. J. Zhang, J. Q. Xu, Z. Shi, X. L. Zhao and T. G. Wang, Chem. Lett., 2002, 31, 1052 CrossRef.
-
V. A. Blatov, TOPOS, A Multipurpose Crystallochemical Analysis with the Program Package, Russia, 2004 Search PubMed.
-
(a) V. A. Blatov and D. M. Proserpio, Acta Crystallogr., Sect. A: Found. Crystallogr., 2009, 65, 202 Search PubMed;
(b) V. A. Blatov, L. Carlucci, G. Giani and D. M. Proserpio, CrystEngComm, 2004, 6, 378 RSC;
(c)
S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, RSC, Cambridge, 2009 Search PubMed;
(d)
L. Öhrström and K. Larsson, Molecule-Based Materials. The Structural Network Approach, Elsevier, Amsterdam, 2005 Search PubMed;
(e) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef CAS;
(f) V. A. Blatov, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 63, 329 CrossRef;
(g)
A. F. Wells, Three-dimensional Nets and Polyhedra, Wiley: New York, ( 1977) Search PubMed;
(h)
A. F. Wells, Further Studies of Three-dimensional Nets, ACA Monograph 8; American Crystallographic Association: Buffalo, NY, 1979 Search PubMed.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- J. Wang, Y. Xu, X. Chen and X. Sun, Compos. Sci. Technol., 2007, 67, 2981 CrossRef CAS.
- Y. K. Zhou, B. L. He, W. J. Zhou, J. Huang, X. H. Li, B. Wu and H. I. Li, Electrochim. Acta, 2004, 49, 257 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.