A simple and robust method for the identification of π–π packing motifs of aromatic compounds†
Leigh
Loots
and
Leonard J.
Barbour
*
Department of Chemistry, University of Stellenbosch, Stellenbosch, 7600, South Africa. E-mail: ljb@sun.ac.za; Fax: +27 21 808-3360; Tel: +27 21 808-3335
First published on 3rd November 2011
Abstract
A simple approach is described for the identification of packing motifs in crystal structures of aromatic molecules using Hirshfeld surfaces and fingerprint plot analysis. The method was first validated by investigating previously studied molecules and establishing trends within these data, and then applied to the analysis of crystal structures of heteroaromatic molecules extracted from the Cambridge Structural Database. A series of cocrystal structures containing these same heteroaromatic molecules were also extracted and investigated using the same method. This simple method appears to be both consistent and robust in distinguishing the different types of packing motifs in structures of aromatic molecules.
Introduction
With regard to structure-directing interactions in the solid-state, much research has been focused on the manner in which various types of hydrogen bonds control molecular packing arrangements. Indeed, these research efforts have now established a hierarchy of supramolecular synthons that can be exploited to control structure, and thus function.1,2 Even in the presence of strong interactions, the organisation of molecules in the solid-state is generally a compromise between both strong and weak interactions. Consequently, the process of predicting crystal structures for any given molecule(s) is extremely challenging.3 The Mercury Materials Module4 of the Cambridge Structural Database5 suite of programs offers valuable tools for identifying most types of hydrogen-bonding motifs in crystal structures, but it is still difficult to recognise packing arrangements that differ even slightly from the reference motif. Moreover, few methods are available to compare π–π interactions and their resulting packing motifs. Thus identifying/classifying packing motifs brought about by weak intermolecular interactions relies largely on the individual researcher's assessment of a structure, which can often be quite subjective.The number of crystal structures being deposited in databases is increasing exponentially. It is thus becoming imperative to develop methods for the rapid recognition, evaluation and comparison of packing arrangements since such phenomena generally govern the properties of crystals. However, some of these property-governing arrangements may be overshadowed by more prominent interactions within the packing arrangement.
π–π interactions in polynuclear aromatic hydrocarbons
Polynuclear aromatic hydrocarbons (PAHs) are well-known to crystallise in four possible structural motifs: herringbone, sandwich-herringbone, γ– or β–structures (Fig. 1).6–8 Each motif is associated with a particular π-interaction geometry. The herringbone motif is constructed from edge-to-face interactions while offset stacking, as well as edge-to-face interactions, are observed in the sandwich herringbone motif where molecules stack in pairs. In the γ–motif, molecules form infinite stacks (offset and/or face-to-face) along one axis and a form of herringbone (edge-to-face) along another axis. The graphitic type layering assembly that arises in the β–motif is promoted primarily by face-to-face or offset stacking of the molecules. Because PAHs consist exclusively of carbon and hydrogen atoms, there are only three possible types of interatomic interactions: C⋯C, H⋯H and C⋯H. The C⋯C interactions are optimised between parallel molecules stacked at van der Waals separation distances, whereas the C⋯H interactions are more effective between inclined molecules.7 It therefore follows that the β– and γ–structures are rich in C⋯C interactions, C⋯H interactions are more prevalent in the herringbone motif, and both types of interactions are observed to a similar extent in the sandwich herringbone arrangement.7 These observations prompted Desiraju and Gavezzotti6–8 to investigate the molecular free surfaces of such molecules and the contributions of molecular areas to the final packing arrangement; atoms located at the core of the molecule, as well as a few peripheral carbon atoms, promote stacking (layering) while the remaining atoms facilitate glide (herringbone) packing.6–8 Desiraju and Gavezzotti went on to say that “it may be expected that whenever similar shapes are found in organic compounds, the effects on crystal structure should be the same, even if the compounds are not pure aromatic hydrocarbons”.7 In a subsequent study, Wilson et al.9,10 were able to confirm this suggestion by comparing the same 32 PAHs by means of Hirshfeld surface11 and fingerprint plot analysis using CrystalExplorer.12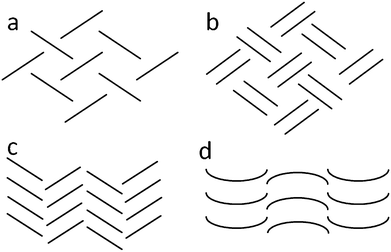 | ||
| Fig. 1 Diagrammatic representation of the a) herringbone, b) sandwich herringbone, c) gamma (γ) and d) beta (β) packing motifs. | ||
Of significance to the present study, Desiraju and Gavezzotti have also proposed that their analysis could be used for heteroatomic derivatives of the PAHs if carbon atoms are substituted with heteroatoms such as N, O, S and P. We have thus extended the work of Desiraju and Gavezzotti,6,7 and Wilson10et al. by considering N-derivatives of benzene, naphthalene and anthracene (Scheme 1). The inclusion of N-atoms would have an influence on the polarisation of the molecules, and therefore the molecular dipoles, and possibly the packing motif.
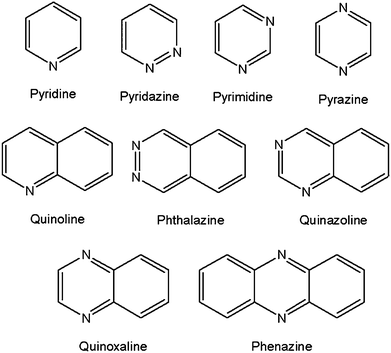 | ||
| Scheme 1 Structural diagrams of the nine N-aromatic compounds selected from the CSD for this study. | ||
π–π interactions in heteroaromatic molecules
In order to evaluate the packing motifs of aromatic molecules in crystals we have used an alternative and more rudimentary method than that of Desiraju and Gavezzotti and Wilson et al. Our approach is similar to that alluded to in 2007 by McKinnon et al.,13 who proposed a quantitative analysis of similar molecules to evaluate their structural similarities. In order to assess the suitability of this method, we first analysed the same 32 PAH structures mentioned above. The general analysis would proceed as follows: obtain crystal structure (experimentally or from the CSD); generate a Hirshfeld surface and fingerprint plot from the CIF file using CrystalExplorer; determine the relative contribution13 of each intermolecular interaction to the Hirshfeld surface (as described in the literature)13 and finally calculate ratios of these interactions (%C⋯H to %C⋯C) for comparison (Fig. 2). These results were then inspected for any discernible trends, which were related back to the actual packing motifs in the crystal structure(s). The herringbone structures all have (%C⋯H to %C⋯C) ratios greater than 4.5, sandwich motifs are in the range of 3.2–4.0; γ–motifs in the range 1.2–2.7 (the single outlier at 7.9 is due to a non-aromatic molecule) and β–motifs fall in the range between 0.46–1.0 (with two outliers) (Fig. 3). (see Table S1 in ESI, †) Following this evaluation of our method against previously investigated data, N–derivatives of benzene, naphthalene and anthracene (Scheme 1) were then extracted from the CSD and analysed in the same manner.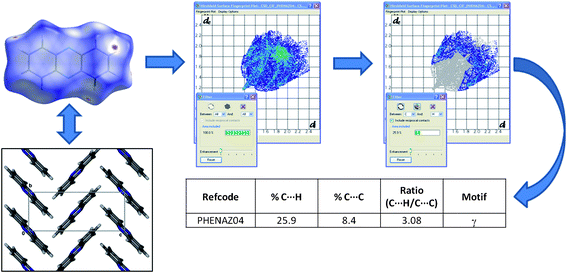 | ||
| Fig. 2 General procedure followed for crystal structure evaluation in the present study. | ||
 | ||
| Fig. 3 Bar chart representation of the ratios of (%C⋯H)/(%C⋯C) interactions and the corresponding packing motifs of PAHs. | ||
The inclusion of nitrogen containing molecules in the study raises the possibility of C–H⋯N interactions occurring – although considered weak interactions, they are generally stronger than the π–interactions being investigated here. Indeed, C–H⋯N interactions, as well as the additional N⋯H, C⋯N and N⋯N contacts, will probably have some effect on the fingerprint plot analyses, but it is still of interest to determine whether the ratio of %C⋯H to %C⋯C can provide important comparative data.
A total of 11 crystal structures (Table 1) fitting our criteria were examined. The structure of pyridine has been reported in two polymorphic forms, both of which crystallise in orthorhombic spacegroups: Form I (PYRDNA0114) in Pna21 and Form II (high pressure, PYRDNA0315) in P212121. Since the shape of the pyridine molecule is similar to that of benzene, we expected that the packing arrangement would be similar as well – and this is true to some extent. Both polymorphs of pyridine are arranged in the herringbone motif, but the herringbone angles are different – a C–H bond of one pyridine molecule is directed more towards the N–atom than towards the centre of its nearest pyridine neighbour. The structures of the diazo-derivatives of benzene (pyridazine, pyrimidine and pyrazine) are all slightly different, but they all pack in the γ–motif. That the packing arrangements are not identical can be expected, given that the molecules are all differently polarised (with respect to each other and to pyridine and benzene). However, the fact that the molecules all opt for the γ–motif rather than the herringbone arrangement is somewhat surprising given their disc-like shape. The above implies that, with a change in polarisation, the diazobenzene molecules are orientated in such a way that π⋯π stacking is favourable, and herringbone interactions between stacks are still possible owing to the small size of the molecules.
| COMPOUND | REFCODE | %C⋯H | %C⋯C | RATIO (%C⋯H)/(%C⋯C) | MOTIF (observed) |
|---|---|---|---|---|---|
| N-Heterocycles | |||||
| Pyridine (Form II) | PYRDNA03 | 32.6 | 0.1 | 326.0 | Herringbone |
| Quinoline | EDAVUA | 39.3 | 0.4 | 98.3 | Herringbone |
| Pyridine (Form I) | PYRDNA01 | 28.4 | 0.5 | 56.8 | Herringbone |
| Phenazine (Form β) | PHENAZ11 | 29.1 | 7.5 | 3.9 | Sandwich |
| Phenazine (Form α) | PHENAZ04 | 25.9 | 8.4 | 3.1 | γ |
| Pyridazine | VOBJEB 22 | 12.1 | 4.2 | 2.9 | γ |
| Phthalazine | DAZNAP | 23.8 | 8.5 | 2.8 | Sandwich |
| Pyrimidine | PRMDIN01 23 | 9.3 | 5.7 | 1.6 | γ |
| Quinazoline | QUINAZ | 14.5 | 9.0 | 1.6 | γ |
| Pyrazine | PYRAZI01 24 | 9.9 | 6.2 | 1.6 | γ |
| Quinoxaline | HEYJOK01 | 10.3 | 11.3 | 0.9 | γ-β |
| Cocrystals | |||||
| Hydroquinone + Pyrazine | QAMQUR | 15.9 | 0.0 | Infinite | Herringbone |
| Hydroquinone + Pyridine | ACESIL | 24.7 | 2.0 | 12.4 | Unknown |
| Hydroquinone + Pyridine | QAMRIG | 20.1 | 10.4 | 1.9 | γ |
| Hydroquinone + Quinoxaline | QEMKAV | 19.2 | 11.1 | 1.7 | γ |
| Hydroquinone + Phenazine | FOQHEY | 18.3 | 15.8 | 1.2 | γ |
Quinoline (EDAVUA16), with a single N–atom, once again assumes a herringbone motif, while the diazanaphthalene derivatives progress from sandwich to γ–motifs as the positions of the nitrogen atoms vary about the ring. An interesting observation within this group of diazanaphthalenes is that the molecules of phthalazine (DAZNAP17) and quinazoline (QUINAZ18) are orientated at 180° relative to each other in forming the offset stacks in the sandwich and γ–motifs. However, the quinoxaline (HEYJOK0119) molecules are simply stacked in a slightly offset manner within the columns.
Phenazine crystallises in two polymorphic forms – Jankowski and Gdaniec20 reported the concomitant crystallisation of both the α–form (PHENAZ0421) and the β–form (PHENAZ1120). Interestingly, although both forms crystallise in P21/n (but with dissimilar unit cell parameters) the α–form packs in the γ–motif, while the β–form (PHENAZ11) takes on the sandwich herringbone motif.
The fingerprint plot analyses of the N-heteroaromatic molecules loosely correlate to those of the PAHs. Although we are not able to predict the packing motifs from these ratios, they do suggest that, with some refinement, this technique could be extremely valuable.
π–π interactions in cocrystals
Using the relatively simple approach described above, we can easily categorise the packing arrangements of pure aromatic substances by focusing on the effects of π–interactions. We were interested in whether the same approach could be used when combining potential hydrogen bond donors with these N-heteroaromatic molecules. Hydrogen-bonded cocrystals would not usually be considered appropriate candidates for the systematic investigation of weak intermolecular interactions, owing to the influence of the much stronger H–bonds; hydrogen bonds often dominate the packing motifs and π–interactions are generally overlooked. To demonstrate the significance of π–interactions in hydrogen bonded networks, a few cocrystal structures from the CSD have been compared. The CSD was interrogated for multi-component structures containing the N-derivatives described above, with a view to finding structures containing a common hydrogen bond donor molecule. Five such structures (see Table 1) were identified, each containing hydroquinone as the hydrogen bond donor – these structures constitute an excellent series since the N–derivatives increase in size and aromaticity. Two distinct forms are obtained by the combination of hydroquinone with pyridine. One form (ACESIL25) crystallises with the asymmetric unit (ASU) consisting of two half molecules of hydroquinone, each situated on an inversion centre, and one molecule of pyridine in a general position. One of the hydroquinone (H) molecules hydrogen bonds to two pyridine (P) molecules to form a P⋯H⋯P heterotrimer, and the remaining hydroquinone acts as a hydrogen bonded bridge between these adducts (Fig. 4a). Although hydrogen bonding appears to play a dominant role in this packing arrangement, a herringbone motif is still discernible, as substantiated by fingerprint plot analysis. In the second form (QAMRIG26) the components combine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of hydroquinone to pyridine, forming the same P⋯H⋯P heterotrimers as described above. However, the hydroquinone hydrogen-bonded bridges are missing – instead, the pyridine molecules stack in an ⋯ABAB⋯ pattern along [001] (Fig. 4b). This arrangement is consistent with the γ–motif, which is also indicated by fingerprint plot analysis.
:2 ratio of hydroquinone to pyridine, forming the same P⋯H⋯P heterotrimers as described above. However, the hydroquinone hydrogen-bonded bridges are missing – instead, the pyridine molecules stack in an ⋯ABAB⋯ pattern along [001] (Fig. 4b). This arrangement is consistent with the γ–motif, which is also indicated by fingerprint plot analysis.
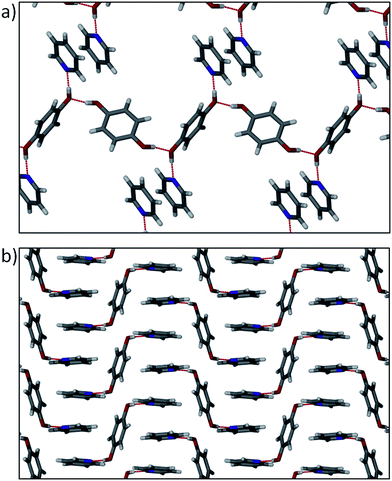 | ||
| Fig. 4 (a) Hydrogen bonded P⋯H⋯P heterotrimers with hydroquinone bridges observed in ACESIL. Some molecules have been omitted for clarity. (b) P⋯H⋯P heterotrimers in QAMRIG showing the pyridine π–π stacking arrangement. | ||
The cocrystal of hydroquinone with pyrazine (QAMQUR26) shows the effect of adding a second N-atom to the acceptor molecule. Both molecules are located on inversion centres and the ASU thus contains only half of each type of molecule. The molecules are connected by means of O–H⋯N hydrogen bonds to form infinite 1–D polymeric chains (Fig. 5). In this structure π⋯π interactions do not seem to play any major role in the arrangement of the molecules.
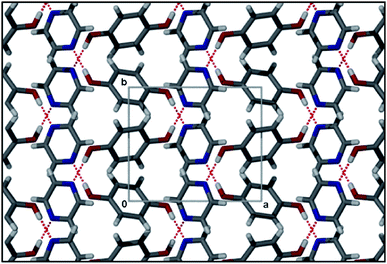 | ||
| Fig. 5 Packing diagram of QAMQUR showing overlapping infinite hydrogren bonded chains. | ||
In the case of hydroquinone/quinoxaline (QEMKAV27), the hydrogen bond acceptor molecule has increased size and aromaticity. This structure crystallises in the triclinic space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with the ASU consisting of half a molecule of hydroquinone located on an inversion centre, and an entire molecule of quinoxaline in a general position. Given the structure containing pyrazine, one might reasonably expect a similar hydrogen bonding pattern and packing motif. However, despite the fact that quinoxaline contains two possible sites for O–H⋯N interactions, only one is utilised in the formation of a heterotrimer. These heterotrimers are stacked by means of π⋯π interactions between the quinoxaline molecules, which are orientated antiparallel to one another (Fig. 6a). The quinoxaline molecules stack in an ⋯ABAB⋯ pattern, with plane separations of 3.388 and 3.487 Å. It is also interesting to compare the hydroquinone/quinoxaline cocrystal structure with that of hydroquinone/phenazine (FOQHEY28), in which half a molecule of hydroquinone and an entire molecule of phenazine are present in the ASU. Hydrogen bonding between these molecules (Fig. 6b) is comparable to that observed in QEMKAV, and the phenazine molecules stack along [100] in an ⋯ABAB⋯ fashion, with plane separations of 3.500 and 3.466 Å. It would be reasonable to assume that H–bonds would be influential in directing molecules into specific packing motifs, and this is what is observed to some extent in the cocrystal structures discussed.
, with the ASU consisting of half a molecule of hydroquinone located on an inversion centre, and an entire molecule of quinoxaline in a general position. Given the structure containing pyrazine, one might reasonably expect a similar hydrogen bonding pattern and packing motif. However, despite the fact that quinoxaline contains two possible sites for O–H⋯N interactions, only one is utilised in the formation of a heterotrimer. These heterotrimers are stacked by means of π⋯π interactions between the quinoxaline molecules, which are orientated antiparallel to one another (Fig. 6a). The quinoxaline molecules stack in an ⋯ABAB⋯ pattern, with plane separations of 3.388 and 3.487 Å. It is also interesting to compare the hydroquinone/quinoxaline cocrystal structure with that of hydroquinone/phenazine (FOQHEY28), in which half a molecule of hydroquinone and an entire molecule of phenazine are present in the ASU. Hydrogen bonding between these molecules (Fig. 6b) is comparable to that observed in QEMKAV, and the phenazine molecules stack along [100] in an ⋯ABAB⋯ fashion, with plane separations of 3.500 and 3.466 Å. It would be reasonable to assume that H–bonds would be influential in directing molecules into specific packing motifs, and this is what is observed to some extent in the cocrystal structures discussed.
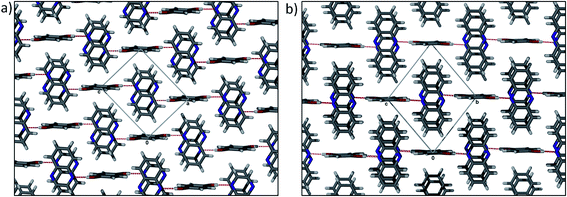 | ||
| Fig. 6 Packing arrangement of (a) hydroquinone/quinoxaline (QEMKAV) and (b) hydroquinone/phenazine (FOQHEY) cocrystals showing the similarities of the two structures. | ||
With limited data on cocrystals of this type (despite the >500,000 structures in the CSD), it is difficult to make any compelling generalisations about the impact of π⋯π interactions on their packing motifs. However, we can conclude that, for this series of cocrystals involving hydroquinone there appears to be a trend towards more pronounced π⋯π interaction with increasing surface area of one of the aromatic molecules. This suggests that there may be a cumulative effect of aromatic interactions that ultimately become strong enough to supplant some of the stronger H–bonds in crystal structures involving aromatic molecules.25
Conclusions
With the vast amount of crystal data deposited in various databases, there is great potential to utilise existing structural information to advance the field of crystal engineering. Indeed, given the rapid expansion of crystal structure analyses, it is becoming increasingly important to develop new methods (or to combine methods) to identify and/or classify observable motifs. The analysis used here has proven to be relatively consistent and robust in identifying the different packing motifs, but still requires some input from the analyst. The simplicity of the method suggests that it will allow rapid comparison of structures and their inherent motifs, although the examples used in this database survey are by no means comprehensive. We believe that the analysis of additional data would yield a more refined estimate of the interaction ratio ranges for the different packing motifs, and that a more sophisticated approach would take heteroatoms into account. For example, based on our limited data we would have predicted a γ motif for phthalazine, which instead assumes a sandwich pattern. However, the %C⋯H to %C⋯C ratio of 2.8 for phthalazine is near the upper limit of the range for γ motifs, and it is reasonable to expect the limits for the different packing motifs to overlap slightly. In this regard we are currently undertaking a more systematic study aimed at expanding the number of cocrystals involving N-aromatic molecules and aromatic alcohols. As yet no theoretical studies have been implemented to corroborate our analysis.Notes and references
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533–4545 CAS.
- G. M. Day, T. G. Cooper, A. J. Cruz-Cabeza, K. E. Hejczyk, H. L. Ammon, S. X. M. Boerrigter, J. S. Tan, R. G. Della Valle, E. Venuti, J. Jose, S. R. Gadre, G. R. Desiraju, T. S. Thakur, B. P. van Eijck, J. C. Facelli, V. E. Bazterra, M. B. Ferraro, D. W. M. Hofmann, M. A. Neumann, F. J. J. Leusen, J. Kendrick, S. L. Price, A. J. Misquitta, P. G. Karamertzanis, G. W. A. Welch, H. A. Scheraga, Y. A. Arnautova, M. U. Schmidt, J. van de Streek, A. K. Wolf and B. Schweizer, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 107–125 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- G. R. Desiraju and A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 473–482 CrossRef.
- G. R. Desiraju and A. Gavezzotti, J. Chem. Soc., Chem. Commun., 1989, 621–623 RSC.
- G. R. Desiraju, Prog. Solid State Chem., 1987, 17, 295–353 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystalExplorer 2.1(381), University of Western Australia, Perth, 2007 Search PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- D. Mootz and H. G. Wussow, J. Chem. Phys., 1981, 75, 1517–1522 CrossRef CAS.
- S. Crawford, M. T. Kirchner, D. Bläser, R. Boese, W. I. F. David, A. Dawson, A. Gehrke, R. M. Ibberson, W. G. Marshall, S. Parsons and O. Yamamuro, Angew. Chem., Int. Ed., 2009, 48, 755–757 CrossRef CAS.
- J. E. Davies and A. D. Bond, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o947–o949 CrossRef.
- C. Huiszoon, W. B. Van Der Waal, A. B. Van Egmond and S. Harkema, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3415–3419 CrossRef CAS.
- C. Huiszoon, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 998 CrossRef.
- S. Ranganathan, S. Mahapatra, T. S. Thakur and G. R. Desiraju, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o2789 Search PubMed.
- W. Jankowski and M. Gdaniec, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o181–o182 CrossRef.
- K. Woźniak, B. Kariuki and W. Jones, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1113–1114 CrossRef.
- A. J. Blake and D. W. H. Rankin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1933 CrossRef.
- S. Furberg, J. Grogaard and B. Smedsrud, Acta Chem. Scand., Ser. B, 1979, 33, 715 CrossRef.
- G.d. With, S. Harkem and D. Feil, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 3178 CrossRef.
- I. D. H. Oswald, W. D. S. Motherwell and S. Parsons, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, O1967–O1969 CrossRef.
- I. D. H. Oswald, W. D. S. Motherwell and S. Parsons, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 46–57 Search PubMed.
- A. Kadzewski and M. Gdaniec, Acta Crystallogr., Sect. E, 2006, 62, o3498 Search PubMed.
- V. R. Thalladi, T. Smolka, R. Boese and R. Sustmann, CrystEngComm, 2000, 2, 96–101 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of fingerprint plot analyses of previously published studies. See DOI: 10.1039/c1ce05763d |
| This journal is © The Royal Society of Chemistry 2012 |