Growth of silver dendritic nanostructuresvia electrochemical route
Mohanrao V.
Mandke
ac,
Sung-Hwan
Han
d and
Habib M.
Pathan
*ab
aAdvanced Physics Laboratory, Department of Physics, University of Pune, Pune, 411007, India. E-mail: pathan@physics.unipune.ac.in; Fax: +91-020-25691684; Tel: +91-020-25692678/25691709 Ext: 323
bCentre for Nanoscience and Quantum System, University of Pune, Pune, 411007, India
cNational Defence Academy, Khadakwasla, Pune, 411023, India
dInorganic Nano-Materials Laboratory, Department of Chemistry, Hanyang University, Haengdang-Dong 17, Seoul, 133-791, Republic of Korea
First published on 10th November 2011
Abstract
Silver dendritic nanostructures were prepared on indium tin oxide coated glass substrate by electrochemical deposition from an aqueous solution of AgNO3 in the presence of citric acid. The silver dendritic nanostructures were characterized by using scanning electron microscopy, X-ray diffraction and UV-Visible absorption spectroscopy. Results showed that the morphology and growth of dendritic structures can be controlled with deposition time. A diffusion-limited aggregation model is used to explain the growth mechanism of silver nanostructures. The UV-Visible absorption spectra showed a wide range of absorption in the visible region by silver dendritic nanostructures.
1. Introduction
Nowadays the synthesis of noble metal nanostructures attracts significant attention and these are being studied extensively by various research groups because of their excellent as well as fascinating optical, catalytic and biosensing properties.1,2 The variety of intrinsic properties the metal nanoparticles offer due to their shape, size, morphology and crystallinity, mean they are candidates suitable for potential applications in the field of optics, microelectronics, catalysis etc.3–7 Therefore, there has been a large interest in the research of synthesis of metal nanostructures with well defined shapes, sizes and crystallinity. Out of the umpteen metal nanoparticle morphologies, the dendritic metal nanostructures have many special characteristics such as a large surface area composed of major trunks and branches. The very good electrical and thermal conductivity of such structures opens up new avenues for their applications in fuel cells8,9 and hydrogen storage devices.10 Among these, silver dendritic nanostructures have become an active research theme because of their potential applications in catalysis11 and in the design of superhybrophobic materials.12Generally metal dendrite structures are observed in non equilibrium growth processes which provides a natural framework for the study of disordered systems.13 Many theoretical and experimental results are reported to study the structure and growth mechanism of various types of dendrites. In this respect many experimental results have been reported to explain the phenomenon of dendritic growth such as the design and growth of dendritic Cu2−xSe crystalline aggregations,14 galvanic replacement mediated growth of dendritic gold nanostructures.15 Recently various experimental techniques have been developed to synthesize the silver dendritic nanostructures such as controllable electrochemical synthesis in AgNO3 and polyvinyl pyrrolidone (PVP) as a surfactant,16 evolution of dendrites in an aqueous solution of AgNO3–HF solution by replacement reaction,17in situ formation of dendritic structures in aqueous solution.18 However, the synthesis of symmetrical, well defined dendritic shaped metal nanostructures is still a challenging field of research.
Among all these techniques, electrodeposition is an effective way for the controlled synthesis of dendritic structures because, in electrodeposition the growth rate of nanostructures on the substrates is the direct function of the applied potential. The applied potential acts as a main driving force for the movement of ions in the electrolyte.
In the present work, probably for the first time the silver dendritic nanostructures consisting of highly ordered trunks, branches and leaves were synthesized via a simple electrodeposition technique in a conventional electrolyte containing an aqueous solution of AgNO3 and citric acid. The growth mechanism of the nanostructure was analyzed on the basis of the DLA model. The optical properties are also analyzed from the UV-Vis absorption spectra.
2. Experimental
Silver nitrate, acetone, ethanol, citric acid were purchased from Thomas Baker. All reagents were used without further treatment. ITO coated glass substrates were cleaned with acetone to remove possible contaminants. The electrolyte prepared contains an aqueous solution of 10 mM AgNO3 and 1M citric acid. The electrodeposition was performed with a two electrode system. ITO coated glass substrate was used as the working electrode and graphite rod as a counter electrode. In a typical synthesis process, the electrodeposition was carried out at 1.5 V at room temperature for the deposition time of 100 s (Sample A), 300 s (Sample B), 600 s (Sample C) and 1200 s (Sample D) in the same solution bath. After deposition, the samples were rinsed with water and dried in air.The morphologies of as-prepared silver samples were characterized by using scanning electron microscopy (SEM, Model No. Jeol-JSM 6360-A). The X-ray diffraction (XRD) patterns were recorded by using an X-ray diffraction analyzer (XRD, Rigaku D/max-2400, Cu-Kα = 0.154 nm). The UV-Vis absorption spectra were collected by using a JascoV-670 spectrometer.
3. Results and analysis
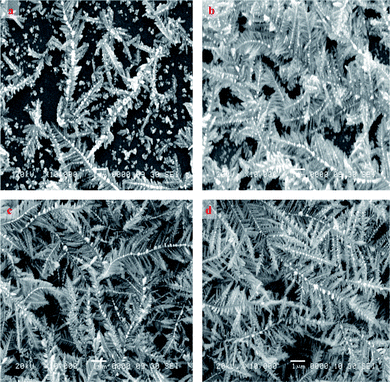
Fig. 1 and 2 represent the typical beautiful SEM images of as-prepared silver dendritic nanostructures for different deposition times at room temperature with two different magnifications. The low magnification images in Fig.1 showed the evolution of silver dendritic nanostructures with time. At the initial stage of electrodeposition some dendritic nuclei of silver were found on the rough surface of the substrate along with a few smaller dendritic nanostructures (Sample A). The apparent number densities of dendritic nuclei and dendritic structures were nearly the same. When the deposition time was raised to 300 s (Sample B), the electric field around the substrate led to the deposition of silver on the surface of already existing particles and nanoparticles start reforming into nanowires at a diameter of about 60–180 nm and a length of about 1.5–2 μm. The applied potential acts as the main driving force for the movement of ions in the electrolyte. Further diffusion of silver ions may lead to the nucleation of silver on the trunk that finally resulted in the formation of dendrite structures. The nanostructures obtained are very thin in the trunk. The diameter of the branches is about 0.04–1.2 μm and their length is about 0.3–1.4 μm. When the deposition time is extended to 600 s (Sample C) and 1200 s (Sample D), the silver dendrite nanostructures grow longer in trunks, branches and leaves along with an increase in branch diameter. | ||
| Fig. 1 Low magnification (3000 ×) SEM images of as-prepared silver dendritic nanostructures for (a) Sample A – 100 s (b) Sample B – 300 s (c) Sample C – 600 s (d) Sample D – 1200 s. | ||
 | ||
| Fig. 2 High magnification (10000 ×) SEM images of as-prepared silver dendritic nanostructures for (a) Sample A – 100 s (b) Sample B – 300 s (c) Sample C – 600 s (d) Sample D – 1200 s. | ||
Further observation of Fig. 2 shows a high magnification of silver dendritic structures with time. Clear, well defined, uniform and more ordered dendrite structures with pronounced trunk and branches uniformly distributed on both sides of the trunk were revealed. It also clearly showed the gradual increase in the number of dendritic structures with deposition time.
A schematic illustration of time dependent growth of individual silver dendrite nanostructures is shown in Fig. 3. The evolution of silver nanostructures was observed to depend upon the reaction time. At the initial stage of deposition, silver nanoparticles of size around 60–160 nm were formed along with some smaller dendritic structures with the appearance of some primary branches (Sample A). A careful observation also showed the appearance of a uniform distribution of nanoparticles on the trunk of silver dendrites. After 300 s (Sample B), a complex morphology with well defined dendritic structures containing primary, secondary and tertiary higher order branches were formed. Also the nanoparticles residing on the trunk started elongating in size in order to form the subsequent branches of silver dendrites. As the reaction progressed, trunks, branches and leaves get more elongated (Sample C and D). Thus, the silver dendrite nanostructure evolved with time from nanoparticles to dendritic structures.
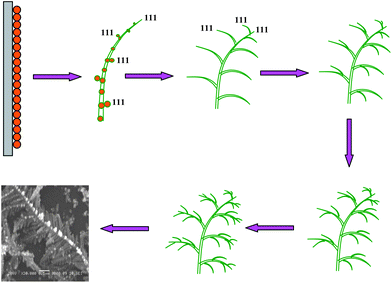 | ||
| Fig. 3 Schematic diagram illustrating the time dependent growth of individual silver dendritic nanostructures. | ||
The growth mechanism of silver dendrite nanostructures may be explained by using diffusion-limited aggregation (DLA) model.19 It provides useful information of the dendritic growth mechanism of silver nanocrystals. This includes a single cluster to which the additional particles get attached once they reach a site adjacent to the edge of the cluster. The additional particles are launched one by one from random positions far away from the cluster and perform a random walk until they either attach to the cluster or move out of the finite system. The rate of nucleation and subsequent growth of dendritic nanostructures are effectively controlled in the reaction.20 In DLA, the growth rate of initially formed nuclei is proportional to 1/R, R being the radius of nuclei. Thus the branches and trunks of individual dendrites become steeper since the smaller nuclei grow rapidly. The Ag2+ nuclei released from random sites perform a random walk until they reach the specific low energy sites on the substrate and get deposited there. The nuclei aggregate successively to form the cluster.
Initially, under the effect of an applied electric field Ag2+ ions accept electrons to form Ag nanoparticles on the working electrode namely the ITO substrate. It is known that the ultimate morphology of the nanoparticles is determined from the different growth rates of crystal faces. As the silver is typical fcc metal, the growth mechanism is more effective along the (111) direction rather than the other directions. So at the initial stage, silver nanoparticles would grow along the (111) direction to form a rod-like silver trunk.
As the reaction continues, the additional silver nanoparticles were synthesized at the same time. The additional particles were launched one by one as suggested in the model above, from random positions and perform a random walk until they either attach to a silver trunk or move out of the finite system. The growth of these attached silver nanoparticles also begins along the (111) direction to form further secondary and tertiary branches. As the reaction proceeded further with time, all the trunks, branches and leaves grew bigger, thicker as well as denser and finally become interconnected to form the most ordered, well oriented silver dendritic nanostructures. As it may be understood from the model, the formation of silver dendritic structures was dominated by both diffusion control and an oriented attachment process.
X-ray diffraction studies revealed that all the samples were crystalline. The typical XRD pattern of as-prepared silver dendrite nanostructures for samples A and B are shown in Fig. 4. All the peaks appearing at 2θ equal to 38.2, 44.3, 64.4 and 77.5 were in agreement with the JCPDS file no. 4-0781 for silver. The peaks at 2θ equal to 50 and 60 were attributed to the substrate namely ITO. This confirmed that the resultant silver dendrites possessed face centered cubic structure. The ratios of intensities of diffraction peaks are calculated from the XRD data and listed in Table 1.
| Sample | Ratio of intensities | |
|---|---|---|
| [111]/[200] | [111]/[[220] | |
| A | 5.64 | 7.52 |
| B | 14.52 | 27.17 |
 | ||
| Fig. 4 XRD patterns of two typical types of as-prepared silver dendritic nanostructures for (a) Sample A – 100 s. (b) Sample B – 300 s. The asterisks represent the XRD pattern of the ITO substrate. | ||
The peak intensity ratio [111]/[200] of samples A and B are 5.64 and 14.52, respectively, which are much higher than the values from a polycrystalline silver with randomly distributed grains (conventional value is around 2.5). The relative intensities of peaks obtained in the XRD hint at the possibility of appearance of dendritic morphology of silver.
It is very interesting to investigate the optical properties of silver since it absorbs strongly in the visible region due to surface plasmon resonance.21 From the results of optical absorption spectra (Fig. 5), it is clear that the shape of the silver nanoparticles has a significant contribution to the absorption peak rather than the size of the nanoparticles. Optical absorption shows comparable results to earlier reported results.22 Usually spherical silver nanoparticles show one size dependent surface plasmon resonance (SPR) absorption band whereas nonspherical geometry such as dendrites show two SPR bands.
 | ||
| Fig. 5 UV-Vis absorption spectra of four typical types of as-prepared silver dendritic nanostructures for (a) Sample A, (b) Sample B, (c) Sample C, (d) Sample D. | ||
All samples have two parts of the absorption peak, one sharp peak of less intensity around 336 nm and another broad peak of high intensity around 454 nm extending over the near infra red region 600–900 nm. The first sharp peak around 336 nm should be attributed to the weak out-of-plane dipole and quadrupole resonance. It is also revealed that the intensity of the wide absorption band increases due to the formation of a large number of highly dense nanoparticles most likely by diffusion-limited-aggregation (DLA).23 As reported before,24 the wide absorption over the near infra red region of the absorption spectrum is due to red shift of relatively intense in-plane dipole resonance where the size distribution and high degree of alignment of the dendritic structure is present.
4. Conclusion
To summarize, the silver dendritic nanostructures have been synthesized on ITO glass substrate by electrodeposition. The advantage of this technique reported here is that the morphology of dendritic nanostructure can be varied with deposition time in addition to applied potential, at constant concentration of AgNO3 and capping agent at room temperature. The growth mechanism of synthesized dendritic nanostructures was explained by the DLA model. The UV-Vis absorption spectrum indicated that the silver dendritic nanostructures have a wide range of absorption in the visible region. These nanostructures may have important applications in catalysis, optics and bioscience, solar cells, etc.Acknowledgements
Authors are thankful to Board of College and University Development, University of Pune, Pune, India and Centre for Nanoscience and Quantum System, University of Pune, Pune, India for their partial financial support. The authors are grateful to the Commandant, Principal and Head, Department of Physics, National Defence Academy, Khadakwasla, Pune, India for their constant co-operation and encouragement.References
- J. Xie, Q. Zhang, J. Y. Lee and D. I. C. Wang, ACS Nano, 2008, 2, 2473 CrossRef CAS.
- W. Ye, C. Shen, J. Tian, C. Wang, L. Bao and H. Gao, Electrochem. Commun., 2008, 10, 625 CrossRef CAS.
- J. Rongchao, Y. C. Cao, H. Encai, G. S. Metraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487 CrossRef.
- W. F. Hoelderich, Catal. Today, 2000, 62, 115 CrossRef CAS.
- W. A. Challener, R. R. Ollmann and K. K. Kam, Sens. Actuators, B, 1999, 56, 254 CrossRef.
- R. Jin, Y. W. Cao, C. A. Mirkin, K. L. Kelly, G. C. Schatz and J. G. Zheng, Science, 2001, 294, 1901 CrossRef CAS.
- I. R. Gould, J. R. Lenhard, A. A. Muenter, S. A. Godleski and S. Farid, J. Am. Chem. Soc., 2000, 122, 11934 CrossRef CAS.
- B. Lim, M. Jiang, P. Camargo, E. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS.
- Z. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542 CrossRef CAS.
- B. D. Adams, G. Wu, S. Nigro and A. Chen, J. Am. Chem. Soc., 2009, 131, 6930 CrossRef CAS.
- M. H. Rashid and T. K. Mandal, J. Phys. Chem. C, 2007, 111, 16750 CAS.
- F. Shi, Y. Song, J. Niu, X. Xia, Z. Wang and X. Zhang, Chem. Mater., 2006, 18, 1365 CrossRef CAS.
- M. Matsushita, M. Sano, Y. Hayakawa, H. Honjo and Y. Sawada, Phys. Rev. Lett., 1984, 53, 286 CrossRef CAS.
- D. Li, Z. Zheng, Y. Lei, S. Ge, Y. Zhang, Yange Zhang, K. W. Wong, F. Yang and W. M. Lau, CrystEngComm, 2010, 12, 1856 RSC.
- X. Bai, Y. Gao and L. Zheng, CrystEngComm, 2011, 13, 3562 RSC.
- X. Hong, G. Z. Wang, Y. Wang, W. Zhu and X. S. Shen, Chin. J. Chem. Phys., 2010, 23, 596 CrossRef CAS.
- W. Ye, C. Shen, J. Tian, C. Wang, C. Hui and H. Gao, Solid State Sci., 2009, 11, 1088 CrossRef CAS.
- W. T. Wu, W. Pang, G. Xu, L. Shi, Q. Zhu, Y. Wang and F. Lu, Nanotechnology, 2005, 16, 2048 CrossRef CAS.
- T. A. Witten Jr and L. M. Sander, Phys. Rev. Lett., 1981, 47, 1400 CrossRef.
- V. Fleury, Nature, 1997, 390, 145 CrossRef CAS.
- S. Kapoor, Langmuir, 1998, 14, 1021 CrossRef CAS.
- J. Zhu, S. Liu, O. Palchik, Y. Koltypin and A. Gedanken, Langmuir, 2000, 16, 6396 CrossRef CAS.
- A. L. Swatek, Z. Dong, J. Shaw Jr and M. Rafiq Islam, J. Exp. Nanosci., 2010, 05, 10 CrossRef CAS.
- Y. Sun and Y. Xia, Adv. Mater., 2003, 15, 695 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |