Layered indium chalcogenidoantimonates [Me2NH2]2In2Sb2S7-xSex (x = 0, 2.20, 4.20, 7) with tunable band gaps and photocatalytic properties†
Kai-Yao
Wang
ab,
Mei-Ling
Feng
a,
De-Nian
Kong
a,
Shi-Jing
Liang
c,
Ling
Wu
c and
Xiao-Ying
Huang
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, the Chinese Academy of Sciences, Fuzhou, Fujian 350002, P.R. China. E-mail: xyhuang@fjirsm.ac.cn; Fax: +86 591 83793727
bGraduate School of the Chinese Academy of Sciences, Beijing, 100049, P. R. China
cState Key Laboratory Breeding Base of Photocatalysis, Research Institute of Photocatalysis, Fuzhou University, Fuzhou, Fujian 350002, P.R. China
First published on 9th November 2011
Abstract
Two novel indium chalcogenidoantimonates and their quaternary mixed solid solutions with a layered structure of [In2Sb2S7-xSex]n2n− are successfully synthesized under mild solvothermal conditions. The compounds show a red-shift of their optical absorption edges and exhibit tunable photocatalytic activity for degradation of methyl orange (MO) with a shift of optical response from UV to the visible light region, as the proportions of Se increase.
As a potential solution to the global energy crisis and environmental problems associated with the emitting of toxic and colored wastewater; the application of photocatalysis for water splitting and elimination of organic pollutants has attracted great attention in the past few decades. Since the use of TiO2 as a catalyst in photochemistry,1 various oxide semiconductor materials have been developed.2 However, most of the photocatalysts can only be active under UV irradiation that takes up less than 5% of the solar energy, which limits their application in energy conversion or environmental remediation to a great extent.
Metal chalcogenides, with suitable energy band gap corresponding to absorption of visible light, are considered to be good candidates for photocatalysis. In the last ten years, multicomponent solid solutions based on metal sulfides have achieved many outstanding results,3 due to their tunable band structures for the most efficient utilization of solar energy. Notably, promising photocatalytic properties have been observed for Cd1−xZnxS,3c, 4 Zn1−xCuxS,5AgInZn7S9,6 ZnIn0.23Ag0.04S1.365,7(CuIn)xCd2(1−x)S2,8AGa2In3S8 (A = Cu or Ag),9a and Cu- or Ni-doped Cd1−xZnxS materials.9b,9c This progress has shown that multicomponent chalcogenides with changeable compositions are a novel class of potential visible-light-driven photocatalysts. However, the systematic study of those photocatalysts with mixed S/Se such as Cu7.2(SexS1−x)4 is far less.10 On the other hand, in contrast to dense solids, open-framework calcogenides are believed to exhibit unique optical and photocatalytic properties because of the possibility of reducing the rate of charge recombination of photogenerated electron-hole pairs.11 Several transition metal-containing sulfides with layered open-frameworks have also been reported for the positive effects their anisotropic structures have on their photocatalytic performance.9a,12 Thus, systematically tuning the structure, as well as the composition of the solid solutions, will be an effective method for designing new photocatalysts.
The incorporation of heterometals into the chalcogenide framework would be likely to enhance the structural diversity with potential properties. The incorporation of group 15 M3+ (M = As, Sb) with active lone pair electrons into conventional metal chalcogenide tetrahedral building units, is attractive because the asymmetric polyhedra MQn (M = As, Sb; Q = S, Se, Te) would likely give rise to novel open-frameworks or even non-centrosymmetric structures with interesting properties.13 As for As3+, some compounds containing AsQ33− and other metal ions, such as Cu+, Hg2+, Ag+, and In3+ have been synthesized.14 When it comes to Sb3+, though many thioantimonates(III) combined with transition metals have been reported,15 the exploration of the thioantimonates(III) with tetrahedral main-group metals such as In3+ and Ga3+ is just in its infancy.16 Moreover, with respect to the above mentioned transition metal sulfides, the microporous or layered crystalline heterometallic chalcogenide materials constructed by main-group metals, to the best of our knowledge, are scarcely reported for photocatalytic processes. Due to their preference for novel open-frameworks or layered structures, main-group heterometallic chalcogenide photocatalysts may represent a brand new working direction, providing us with a large area to explore.
Recently, we have dedicated much effort to searching for new heterometallic chalcogenides by combining group 13 (or 14) with group 15 elements.13a, 16c, 17 To aid continuity and further development of our research, we herein report on the syntheses, structures, and optical properties of four isomorphic indium chalcogenidoantimonates, namely, [(Me)2NH2]2[In2Sb2S7−xSex] (x = 0 (1), 2.20 (2), 4.20 (3), and 7 (4)), among which 4 is especially the first example of indium selenidoantimonate induced by organic amines. Significantly, the compounds show a red-shift of their optical absorption edges and expand the photocatalytic activity for degradation of methyl orange (MO) in water solution from UV to the visible light region, as the proportions of Se increase, demonstrating an excellent example of composition-dependent optical property and photocatalytic performance.
Lamellar crystals of 1–4 were all solvothermally synthesized by reacting In2S3, Sb2S3 (or elements In and Sb for 4) with S or Se in a mixed solvent of N,N′-dimethylformamide (DMF) and hydrazine hydrate (for details, see ESI†). As reported by ourselves recently,13a, 16c, 18 the DMF, not only as solvent, can hydrolyze in situ to produce dimethylammonium ions, which enter the structures of the compounds as template and charge-balancing agents. Though we can not clearly figure out how the addition of hydrazine hydrate influences the reactions, the volume proportion of the two solvents is crucial for the reactions. In the absence of hydrazine hydrate, only indefinite brown and black powders with a few red particles were formed in similar reactions for 1 and 4, respectively. Only by tuning the proportions (DMF to hydrazine hydrate) to 3.75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1, 5:1; 6:1, could we obtain compounds 1–4 in the highest yields, respectively.
:1, 5:1, 5:1; 6:1, could we obtain compounds 1–4 in the highest yields, respectively.
Single crystal X-ray analysis reveals that 1, 2, 3 and 4 are isomorphic, belonging to space groupC2/c.‡ Their structures feature organic amine intercalated inorganic anionic layers of [In2Sb2S7-xSex]n2n−. Therefore, only the structure of 4 will be described in detail (Fig. 1), on behalf of a novel In/Sb/Se composition. The asymmetric unit of 4 consists of one In3+ ion, one Sb3+ ion, half a Se2− (Se1) ion with 2 symmetry, three other Se2− ions, and one [(Me)2NH2]+ cation. The In3+ ion is surrounded by four Se2− to form a distorted tetrahedron, in which the In–Se lengths range from 2.5663(6) to 2.5840(8) Å, comparable to those reported.19 The Sb3+ ion adopts a slightly distorted trigonal-pyramidal coordination geometry, where the interatomic distances and angles lie in the range of those in similar structures.20 All Se2− ions act as bidentate metal linkers, among which Se1 bridges two In3+ ions, and the remaining Se2− ions link one In3+ ion and one Sb3+ ion, respectively. The alternating connection of two InSe4 and two SbSe3 through Se(2) and Se(3) vertexes gives rise to a tetranuclear {In2Sb2Se10} cluster (Fig. 1a). Though similar metal thionate structural units have been observed in [Mn(tren)]4Mn2Sb4S12,21[Fe(tren)]FeSbS4,22 [La(en)4SbSnS5]2·0.5H2O,17[Mn(tren)]InAsS4,14d and [M(dap)3]InSb3S7 (M = Co, Ni),16e this type of unit containing selenium is the first reported here. The neighboring clusters interlink to each other through two Se(4)2− ions via corner-sharing to form an infinite [In2Sb2Se8]n4n− ribbon running along the [010] direction. This connection mode differs entirely from those in 1D [FeSbS43−]n, [InAsS42−]n and [In2Sb2S84−]n in the above-mentioned compounds, in which the neighboring rings are linked through single metal ion vertexes (Fig. S2†). Then, the adjacent [In2Sb2Se8]n4n− ribbons are further weaved into a 2D anionic layer of [In2Sb2Se7]n2n− in an anti-parallel fashion by using corner-shared Se1 atoms (Fig. 1b). Accompanied by the formation of a layer, an arrow-like six-membered ring of [In4Sb2Se6] can be observed in the interspace of two ribbons. This ring, composed of four InSe4 tetrahedra and two SbS3 trigonal pyramids, represents a new linkage sequence in Ga/In-As/Sb-Q (Q = S, Se, Te) system. The layers stack in an AAA sequence along the a axis with an interlayer spacing of about 4.63 Å. The protonated dimethylammonium ions as templates and charge-balancing agents are sandwiched in the inter-layer spaces. A hydrogen-bond relationship is proposed as the extensive N–H⋯Se and C–H⋯Se bond distances and angles lie in the range of 3.474(6)–3.882(8) Å and 144.4–173.3°, respectively (Table S3†). The H-bonding interactions between the cations and anionic layers results in a 3D network (Fig.1c).
![(a) Tetranuclear heterometallic cluster of {In2Sb2Se10} in 4; (b) [In2Sb2Se7]n2n− layer in 4; (c) perspective view of packing of 4 along the b axis. Dotted lines show the N–H⋯Se and C–H⋯Se hydrogen bonds.](/image/article/2012/CE/c1ce05879g/c1ce05879g-f1.gif) | ||
| Fig. 1 (a) Tetranuclear heterometallic cluster of {In2Sb2Se10} in 4; (b) [In2Sb2Se7]n2n− layer in 4; (c) perspective view of packing of 4 along the b axis. Dotted lines show the N–H⋯Se and C–H⋯Se hydrogen bonds. | ||
When the Se atoms in 4 are completely or partially substituted by S atoms, the compounds 1, 2 and 3 form. Energy dispersive X-ray spectroscopy (EDS) analyses on 2 and 3 confirm the presence of both S and Se in the compounds (Fig. S3†). In company with the results of elemental analyses and crystal structure refinements, the final formulae are determined as [(Me)2NH2]2[In2Sb2S4.80Se2.20] for 2 and [(Me)2NH2]2[In2Sb2S2.80Se4.20] for 3, respectively. As shown in the inset of Fig. 2a, the (200) peaks in the powder X-ray diffraction (PXRD) pattern shift from 10.72° in 1 to 10.38° in 4 as larger Se ions substitute more S ions when x increases. The composition dependence of the unit cell volumes is displayed in Fig. 2b, where the volumes show an increase with the introduction of the Se into the sulfide structure, following Vegard's law of solid solution. A total volume increase from x = 0 to x = 7 amounts to 162.5 Å3, i.e. about 8.41%. In this figure, we can also observe the linear evolution of the lattice parameters as a function of Se content. Especially, these increases are most notable for the a axis, which may be due to a larger atom radius and lower electronegativity of Se to form weaker H-bonds, in contrast to those of S atoms.
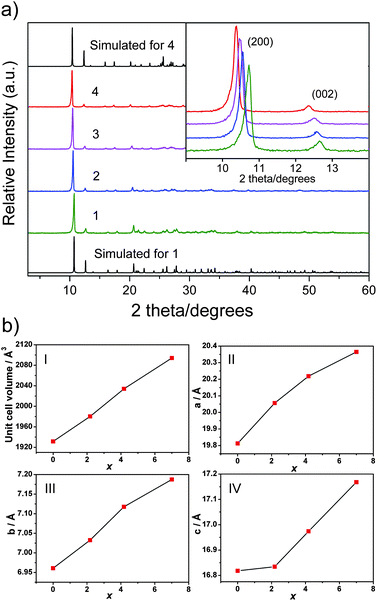 | ||
| Fig. 2 (a) PXRD patterns of compounds 1–4; (b) changes of the unit cell volumes (I), cell constants a (II), b (III) and c (IV) versus x. | ||
Thermogravimetric analyses (TGA) were performed on the pure powder samples of the title compounds in a N2 atmosphere (Fig. S4†). Their TGA curves show that all the compounds are stable up to 210 °C. The main steps of weight losses of 15.69%, 14.41%, 12.49% occur in the range of 210–390 °C for 1, 2, and 3, respectively, assigned to the removal of two dimethylamine molecules and one H2S molecule per formula (calcd. 15.73%, 13.91%, 12.59% for 1, 2 and 3). However, compound 4 has a total weight loss of 14.91% just reaching 310 °C, which can be attributed to the loss of the organic amines and one molecule of H2Se, close to the theoretical value of 15.31%.
UV-Vis optical diffuse-reflectance spectra of the end-members and solid solutions are plotted in Fig. 3. The band gaps of 1–4, were estimated to be 2.31, 1.93, 1.78 and 1.61 eV, respectively. Compared with the bulk In2S3 (2.3 eV),23Sb2S3 (1.6 eV),24In2Se3 (1.36 eV)25 and Sb2Se3 (1.21 eV),26 the absorption edges of the end-members 1 and 4 exhibit a blue-shift with respect to their counterparts. In addition, the curves clearly demonstrate that the absorption edges for the series of [(Me)2NH2]2[In2Sb2S7−xSex] monotonically red shift as the Se mole fraction (x) increases. And it is worth noting that the absorption edge differential as large as 0.70 eV between 1 and 4 represents an effective method to tune the band gap of the materials by changing their compositions. Absorption edges of all four compounds lie in the energy range corresponding to visible light, which are suitable for photocatalytic investigations.11
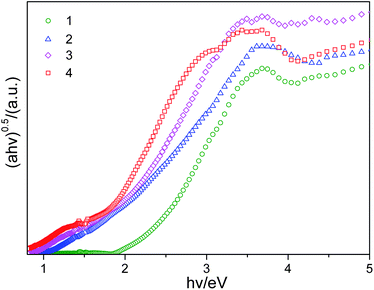 | ||
| Fig. 3 Variation of (αhυ)1/2 as a function of photon energy for 1, 2, 3 and 4. | ||
Comparative experiments were carried out to demonstrate the potential application of the as-prepared [(Me)2NH2]2[In2Sb2S7−xSex] samples in the photodegradation of organic contaminant under UV and visible light irradiation, by choosing methyl orange (MO) as a test pollutant. Temporal changes in the concentration of MO were monitored by examining the variations of the intensities in the maximal absorption in UV-Vis spectra at about 464 nm. Fig. 4a shows the results of MO photodecolorization over the samples under UV light illumination. It is obvious that the photocatalytic activities of the sulfide is higher than that of the samples containing Se under UV light irradiation. The degradation ratio of MO over sample 1 reached 96.8% after 3 h and it was evaluated to be about 74.5% for x = 2.20, 85.1% for x = 4.20, and 78.2% for x = 7. On the other hand, the phenomenon under visible light shown in Fig. 4b is different. In general, the more Se content it contains, the higher the efficiency of the photocatalytic activity can be expected to be. After 10 h of irradiation, degradation of MO was not observed without photocatalyst, while that in the presence of [(Me)2NH2]2[In2Sb2S7−xSex] solid solution were 18.3%, 11.5%, 51.9% and 91.5% when x values are equal to 0, 2.20, 4.20, and 7, respectively. Leaving out the induction periods, the obvious shift of photocatalysis from UV light response to visible light response is likely related to the change of the band structures of the solid solutions. Increasing of the Se content results in a red shift of the absorption edge, which can help the sample to absorb visible light in a wider range of wavelength, and thus enhance the photocatalytic activity. A photocatalytic reaction for degrading MO over sample 4 was also carried out under 600–780 nm light irradiation which cannot be absorbed by the MO molecule (Fig. S7†). Compared to the degradation results as shown in Fig. 4b, the degradation efficient is lower, implying that a significant portion of MO might be degraded through the “indirect photocatalytic mechanism”27 under visible light (400–800 nm) irradiation. In addition, the photocatalytic activities of the catalysts under visible light have also been evaluated by decomposing the colorless substrates salicylic acid and 4-chlorophenol. However, both were not degraded at all. XRD characterizations demonstrate that the compounds are essentially stable after the photocatalytic processes (Fig. S8†). The total organic carbon (TOC) value of the solution rises from 5.48 mg L−1 to 12.56 mg L−1 after 5 h UV irradiation over sample 1, which may be caused by the inevitable diffusion of the small amount of catalyst particles or organic amines into the solution tested. Thus it is not clear whether a simple decoloration or complete mineralization occurred in this case.
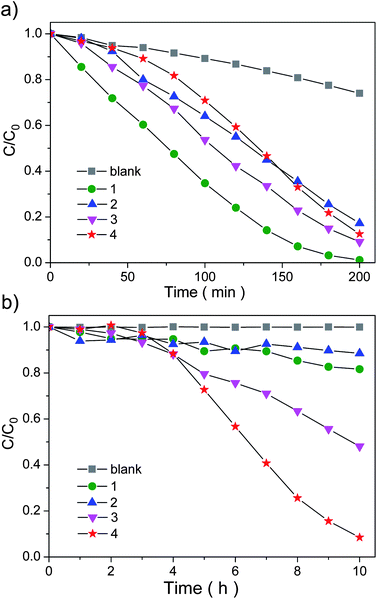 | ||
| Fig. 4 Photodegradation of MO monitored as the normalized change in concentration as a function of irradiation time under (a) UV light and (b) visible light. | ||
In conclusion, the syntheses of a novel series of quarternary main-group metallic chalcogenide solid solutions directed by in situ generated organic amines, namely, [(Me)2NH2]2[In2Sb2S7−xSex] is our initial step to study the correlation between the composition and properties of heterometallic chalcogenides. All these materials features organic amine templated inorganic anionic layer of [In2Sb2S7−xSex]n2n−, representing a new combination of InQ4 tetrahedra and SbQ3 trigonal pyramids. In particular, 4 is the first example of indium selenidoantimonate containing an organic component. The band gap energies can be narrowed by increasing the Se content in the compounds, suggesting the possibility of changing the optical and electronic properties by tuning the S/Se ratio systematically. Furthermore, experimental MO photodegradation demonstrates the increase of the Se content can expand photocatalytic activity from UV to visible light region, which is in agreement with their tunable absorption edges. Undoubtedly, using the synthetic strategy applied here, as we anticipate, plenty of new main-group heterometallic chalcogenide photocatalysts with structural diversity can be developed.
Acknowledgements
This work was supported by the Knowledge Innovation Program of the Chinese Academy of Sciences (KJCX2-YW-H21), the NNSF of China (Grants 20771102, 20873149, 20803081), and the NSF of Fujian Province (No. 2008J0174, 2010J01056).Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- (a) M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS; (b) M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505–516 CrossRef CAS; (c) M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC; (d) D. S. Bhatkhande, V. G. Pangarkar and A. A. C. M. Beenackers, J. Chem. Technol. Biotechnol., 2002, 77, 102–116 CrossRef CAS.
- (a) I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406–13413 CrossRef CAS; (b) I. Tsuji, H. Kato and A. Kudo, Angew. Chem., Int. Ed., 2005, 44, 3565–3568 CrossRef CAS; (c) L. Wang, W. Z. Wang, M. Shang, W. Z. Yin, S. M. Sun and L. Zhang, Int. J. Hydrogen Energy, 2010, 35, 19–25 CrossRef CAS.
- (a) K. Zhang, D. W. Jing, C. J. Xing and L. J. Guo, Int. J. Hydrogen Energy, 2007, 32, 4685–4691 CrossRef CAS; (b) J. Chen, S. Lin, G. Y. Yan, L. Y. Yang and X. Q. Chen, Catal. Commun., 2008, 9, 65–69 CrossRef CAS; (c) W. Z. Wang, W. Zhu and H. L. Xu, J. Phys. Chem. C, 2008, 112, 16754–16758 CrossRef CAS; (d) J. G. Yu, J. Zhang and M. Jaroniec, Green Chem., 2010, 12, 1611–1614 RSC; (e) X. Xu, R. J. Lu, X. F. Zhao, S. L. Xu, X. D. Lei, F. Z. Zhang and D. G. Evans, Appl. Catal., B, 2011, 102, 147–156 CrossRef CAS.
- A. Kudo and M. Sekizawa, Catal. Lett., 1999, 58, 241–243 CrossRef CAS.
- A. Kudo, I. Tsuji and H. Kato, Chem. Commun., 2002, 1958–1959 RSC.
- Y. X. Li, G. Chen, C. Zhou and J. X. Sun, Chem. Commun., 2009, 2020–2022 RSC.
- L. Ren, F. Yang, Y. R. Deng, N. N. Yan, S. Huang, D. Lei, Q. Sun and Y. Yu, Int. J. Hydrogen Energy, 2010, 35, 3297–3305 CrossRef CAS.
- (a) H. Kaga, K. Saito and A. Kudo, Chem. Commun., 2010, 46, 3779–3781 RSC; (b) G. J. Liu, L. Zhao, L. J. Ma and L. J. Guo, Catal. Commun., 2008, 9, 126–130 CrossRef CAS; (c) X. H. Zhang, D. W. Jing, M. C. Liu and L. J. Guo, Catal. Commun., 2008, 9, 1720–1724 CrossRef CAS.
- Y. H. Gao, Z. Zheng, F. L. Yang, F. J. Zhang, P. J. Li, W. J. Fa, H. M. Jia and H. X. Zhao, CrystEngComm, 2011, 13, 1441–1445 RSC.
- N. F. Zheng, X. H. Bu, H. Vu and P. Y. Feng, Angew. Chem., Int. Ed., 2005, 44, 5299–5303 CrossRef CAS.
- S. H. Shen, L. Zhao and L. J. Guo, J. Phys. Chem. Solids, 2008, 69, 2426–2432 CrossRef CAS.
- (a) M. L. Feng, D. N. Kong, Z. L. Xie and X. Y. Huang, Angew. Chem., Int. Ed., 2008, 47, 8623–8626 CrossRef CAS; (b) N. Ye, Q. X. Chen, B. C. Wu and C. T. Chen, J. Appl. Phys., 1998, 84, 555–558 CrossRef CAS; (c) P. S. Halasyamani and K. R. Poeppelmeier, Chem. Mater., 1998, 10, 2753–2769 CrossRef CAS; (d) T. K. Bera, J. H. Song, A. J. Freeman, J. I. Jang, J. B. Ketterson and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2008, 47, 7828–7832 CrossRef CAS; (e) J. L. Mertz, N. Ding and M. G. Kanatzidis, Inorg. Chem., 2009, 48, 10898–10900 CrossRef CAS; (f) T. K. Bera, J. I. Jang, J. B. Ketterson and M. G. Kanatzidis, J. Am. Chem. Soc., 2009, 131, 75–77 CrossRef CAS.
- (a) J. E. Jerome, P. T. Wood, W. T. Pennington and J. W. Kolis, Inorg. Chem., 1994, 33, 1733–1734 CrossRef CAS; (b) J. H. Chou and M. G. Kanatzidis, Chem. Mater., 1995, 7, 5–8 CrossRef CAS; (c) M. Wachhold and M. G. Kanatzidis, Inorg. Chem., 1999, 38, 3863–3870 CrossRef CAS; (d) Z. Q. Wang, H. J. Zhang and C. Wang, Inorg. Chem., 2009, 48, 8180–8185 CrossRef CAS.
- (a) H. O. Stephan and M. G. Kanatzidis, J. Am. Chem. Soc., 1996, 118, 12226–12227 CrossRef CAS; (b) G. L. Schimek, W. T. Pennington, P. T. Wood and J. W. Kolis, J. Solid State Chem., 1996, 123, 277–284 CrossRef; (c) P. Vaqueiro, A. M. Chippindale, A. R. Cowley and A. V. Powell, Inorg. Chem., 2003, 42, 7846–7851 CrossRef CAS; (d) V. Spetzler, C. Näther and W. Bensch, Inorg. Chem., 2005, 44, 5805–5812 CrossRef CAS; (e) D. N. Kong, Z. L. Xie, M. L. Feng, D. Ye, K. Z. Du, J. R. Li and X. Y. Huang, Cryst. Growth Des., 2010, 10, 1364–1372 CrossRef CAS.
- (a) N. Ding and M. G. Kanatzidis, Nat. Chem., 2010, 2, 187–191 CrossRef CAS; (b) N. Ding and M. G. Kanatzidis, Chem. Mater., 2007, 19, 3867–3869 CrossRef CAS; (c) M. L. Feng, Z. L. Xie and X. Y. Huang, Inorg. Chem., 2009, 48, 3904–3906 CrossRef CAS; (d) J. Zhou, X. H. Yin and F. Zhang, Inorg. Chem., 2010, 49, 9671–9676 CrossRef CAS; (e) J. Zhou, L. T. An and F. Zhang, Inorg. Chem., 2011, 50, 415–417 CrossRef CAS; (f) X. Liu, Inorg. Chem. Commun., 2011, 14, 437–439 CrossRef CAS; (g) J. Zhou and L. T. An, CrystEngComm, 2011, 13, 5924–5928 RSC.
- M. L. Feng, D. Ye and X. Y. Huang, Inorg. Chem., 2009, 48, 8060–8062 CrossRef CAS.
- J. R. Li and X. Y. Huang, Dalton Trans., 2011, 40, 4387–4390 RSC.
- (a) C. Wang, X. H. Bu, N. F. Zheng and P. Y. Feng, Chem. Commun., 2002, 1344–1345 RSC; (b) M. J. Manos, C. D. Malliakas and M. G. Kanatzidis, Chem.–Eur. J., 2007, 13, 51–58 CrossRef; (c) P. Vaqueiro, Inorg. Chem., 2008, 47, 20–22 CrossRef CAS.
- (a) T. M. Martin, P. T. Wood and J. W. Kolis, Inorg. Chem., 1994, 33, 1587–1588 CrossRef CAS; (b) D. M. Smith, C.-W. Park and J. A. Ibers, Inorg. Chem., 1996, 35, 6682–6687 CrossRef CAS; (c) D. X. Jia, Y. Zhang, Q. X. Zhao and J. Deng, Inorg. Chem., 2006, 45, 9812–9817 CrossRef CAS.
- M. Schaefer, C. Näther and W. Bensch, Solid State Sci., 2003, 5, 1135–1139 CrossRef CAS.
- R. Kiebach, W. Bensch, R.-D. Hoffmann and R. Pöttgen, Z. Anorg. Allg. Chem., 2003, 629, 532–538 CrossRef CAS.
- T. Asikainen, M. Ritala and M. Leskelä, Appl. Surf. Sci., 1994, 82–83, 122–125 CrossRef.
- T. Fujita, K. Kurita, K. Tokiyama and T. Oda, J. Phys. Soc. Jpn., 1987, 56, 3734–3739 CrossRef CAS.
- C. Julien, A. Chevy and D. Siapkas, Phys. Status Solidi A, 1990, 118, 553–559 CrossRef CAS.
- R. Vadapoo, S. Krishnan, H. Yilmaz and C. Marin, Phys. Status Solidi B, 2011, 248, 700–705 CrossRef CAS.
- (a) B. Ohtani, Chem. Lett., 2008, 37, 216–229 CrossRef; (b) B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS; (c) C. C. Chen, W. H. Ma and J. C. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC; (d) J. Ryu and W. Choi, Environ. Sci. Technol., 2008, 42, 294–300 CrossRef CAS; (e) H. Kisch and W. Macyk, ChemPhysChem, 2002, 3, 399–400 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses, crystallographic data, selected bond lengths and angles table, hydrogen bond table, TGA, PXRD of residues after TGA and EDS results. CCDC reference numbers 832588–832591. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce05879g |
| ‡ Crystal and structure refinement parameters for 1: C4H16In2N2S7Sb2, λ = 0.71073 Å, Mr = 789.75, monoclinic, C2/c, a = 19.8124(6), b = 6.96070(10), c = 16.8179(4) Å, β = 123.610(17)°, V = 1931.6(4) Å3, Z = 4, T = 296(2)K, ρc = 2.716 g cm−3, μ(Mo-Kα) = 5.864 mm−1, 7084 reflections collected, 2217 independent reflections. R1 = 0.0338, wR2 = 0.0833 [I > 2σ(I)]. For 2: C4H16In2N2S4.80Sb2Se2.20, λ = 0.71073 Å, Mr = 892.93, monoclinic, C2/c, a = 20.0561(16), b = 7.0325(3), c = 16.8343(16) Å, β = 123.485(12)°, V = 1980.3(4) Å3, Z = 4, T = 296(2)K, ρc = 2.995 g cm−3, μ(Mo-Kα) = 9.532 mm−1, 6427 reflections collected, 2097 independent reflections. R1 = 0.0471, wR2 = 0.1212 [I > 2σ(I)]. For 3: C4H16In2N2S2.80Sb2Se4.20, λ = 0.71073 Å, Mr = 986.73, monoclinic, C2/c, a = 20.2182(19), b = 7.1175(3), c = 16.974(3) Å, β = 123.619(14)°, V = 2034.1(5) Å3, Z = 4, T = 296(2)K, ρc = 3.222 g cm−3, μ(Mo-Kα) = 12.654 mm−1, 4660 reflections collected, 2146 independent reflections. R1 = 0.0390, wR2 = 0.0596 [I > 2σ(I)]. For 4, C4H16In2N2Sb2Se7, λ = 0.71073 Å, Mr = 1118.05, monoclinic, C2/c, a = 20.3648(11), b = 7.1872(2), c = 17.1677(15) Å, β = 123.551(9)°, V = 2094.1(2) Å3, Z = 4, T = 296(2)K, ρc = 3.546 g cm−3, μ(Mo-Kα) = 16.879 mm−1, 3977 reflections collected, 2162 independent reflections. R1 = 0.0253, wR2 = 0.0407 [I > 2σ(I)]. |
| This journal is © The Royal Society of Chemistry 2012 |