DOI:
10.1039/C1CE05441D
(Paper)
CrystEngComm, 2012,
14, 200-210
Evaluation of the role of disordered organic fluorine in crystal packing: insights from halogen substituted benzanilides†
Received
13th April 2011
, Accepted 24th August 2011
First published on 24th October 2011
Abstract
The determination of the crystal and molecular structures of a large number of compounds containing the C(sp2)-F bond has been investigated in detail in halogenated benzanilides and also in liquids, namely the fluorinated amines. It has been observed that when the fluorine atom is present in the ortho or meta position with respect to the amide functionality in benzanilides or the amino group in fluorinated amines which are liquids at room temperature, the fluorine atom exhibits positional disorder. This is associated with changes in patterns of intermolecular interactions which affect crystal packing. Furthermore, the presence of a fluorine atom on the benzanilide framework, in the presence of a heavier halogen (chloro, bromo and iodo), meta or ortho to the amide group does not eliminate the disorder associated with these molecules. In this article, we highlight the salient features present in halogenated compounds exhibiting disorder in the position of organic fluorine with concomitant changes in crystal packing. This feature is also compared with related compounds exhibiting similarity in electronic features, namely positional disorder.
Introduction
The C–F bond is one of the strongest bonds known in the chemical literature.1 It is of immense importance from a synthetic perspective and also its implications in bio-molecules have been widely recognized.2 Of significance is the supramolecular chemistry delineating the role of organic fluorine in crystal engineering.3 There have been detailed studies exploring the possibility of formation of intermolecular interactions of the type C–H⋯F, F⋯F in different organic molecules containing organic fluorine in addition to the formation of the well known hydrogen bonds.4
In this regard, a detailed crystallographic investigation performed on halogen substituted benzanilides has brought out the significance of strong N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–H⋯OC and C–H⋯F–C hydrogen bonds acting in conjunction with weak C–H⋯π interactions along with the presence of homo-halogen X⋯X and X⋯Y interactions, where X, Y: –F, –Cl, –Br, –I.5 The crystal and molecular structures investigated exhibited no positional disorder of the halogen atom.5 However, structural determinations of compounds containing the lightest halogen atom, namely fluorine, indicates that the fluorine atom tends to get disordered over different positions in the molecule, particularly when the fluorine atom is present in an ortho or meta position with any given functional group in an organic molecule.6 This feature has also been observed with mono- and difluorinated benzanilides.7 This has also resulted in the formation of polymorphs in 3-fluoro-N-(3-fluorophenyl)benzamide, wherein the positional disorder associated with the fluorine atom generates different packing motifs, stabilized by C–H⋯F and F⋯F contacts resulting in changes in crystal packing.8 Furthermore, there exist differences in the magnitude of positional disorder in the unsolvated and solvated forms of 3-fluorobenzoylaminophenyl-3-fluorobenzoate generating different packing motifs involving C–H⋯F interactions in the crystal lattice.9 Another interesting aspect related to this disorder is observed in 2-fluoro aniline, leading to a high Z′ structure.10 In all cases intermolecular interactions like C–H⋯F and F⋯F have been observed. It is also to be noted that such a disorder brings in intermolecular halogen⋯halogen contacts more prominently as the position of the fluorine atom is at two sites, one with a higher occupancy and the other with lower occupancy. It is desirable therefore to develop a protocol to handle the refinement of disorder keeping in view the possible presence of intermolecular halogen⋯halogen contacts. It is also of importance to analyze the propensity of formation of fluorine involved interactions with respect to site occupancy. In one such case i.e. in 4-(2-fluoro-phenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-pyrimidine-5-carboxylic acid ethyl ester, it was observed that the initial structure determination was performed without treatment of disorder of the orthofluorine which resulted in the value of high residual electron density.11 Subsequently, the redetermination was performed again wherein it was observed that the fluoro atom is disordered over two sites, the final refinement giving an acceptable value of the residuals in electron density.12
C, C–H⋯OC and C–H⋯F–C hydrogen bonds acting in conjunction with weak C–H⋯π interactions along with the presence of homo-halogen X⋯X and X⋯Y interactions, where X, Y: –F, –Cl, –Br, –I.5 The crystal and molecular structures investigated exhibited no positional disorder of the halogen atom.5 However, structural determinations of compounds containing the lightest halogen atom, namely fluorine, indicates that the fluorine atom tends to get disordered over different positions in the molecule, particularly when the fluorine atom is present in an ortho or meta position with any given functional group in an organic molecule.6 This feature has also been observed with mono- and difluorinated benzanilides.7 This has also resulted in the formation of polymorphs in 3-fluoro-N-(3-fluorophenyl)benzamide, wherein the positional disorder associated with the fluorine atom generates different packing motifs, stabilized by C–H⋯F and F⋯F contacts resulting in changes in crystal packing.8 Furthermore, there exist differences in the magnitude of positional disorder in the unsolvated and solvated forms of 3-fluorobenzoylaminophenyl-3-fluorobenzoate generating different packing motifs involving C–H⋯F interactions in the crystal lattice.9 Another interesting aspect related to this disorder is observed in 2-fluoro aniline, leading to a high Z′ structure.10 In all cases intermolecular interactions like C–H⋯F and F⋯F have been observed. It is also to be noted that such a disorder brings in intermolecular halogen⋯halogen contacts more prominently as the position of the fluorine atom is at two sites, one with a higher occupancy and the other with lower occupancy. It is desirable therefore to develop a protocol to handle the refinement of disorder keeping in view the possible presence of intermolecular halogen⋯halogen contacts. It is also of importance to analyze the propensity of formation of fluorine involved interactions with respect to site occupancy. In one such case i.e. in 4-(2-fluoro-phenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-pyrimidine-5-carboxylic acid ethyl ester, it was observed that the initial structure determination was performed without treatment of disorder of the orthofluorine which resulted in the value of high residual electron density.11 Subsequently, the redetermination was performed again wherein it was observed that the fluoro atom is disordered over two sites, the final refinement giving an acceptable value of the residuals in electron density.12
Furthermore, the presence of symmetry in the molecule has been shown to influence disorder in the fluorine position, for example, in 2-fluorobiphenyl, trans-4-fluoroazoxy-benzene and trans-4-fluoro-NNO-azoxybenzene.13,14 The molecular assemblies in these structures have been shown to be stabilized by C–H⋯F hydrogen bonds. The question remains whether the intermolecular interactions are observed as a consequence of geometry alone. In fact, the C–F bond length associated with the major occupancy fluorine is almost close to the standard C–F bond length (1.345 Å), while the minor occupancy fluorine has a bond length lying between normal values of C–H and C–F bond.14
Several of the possible combinations of fluorine substitution both on the aniline side and on the benzoyl side show disorder in fluorine positions. This article is mainly devoted to the discussion related to positional disorder in fluorine in halogen substituted benzanilides. The structures to be discussed in this section provide an opportunity to evaluate the features of intermolecular interactions amongst disordered atoms (Scheme 1).
![Schematic representation [nnXA–nnYB; n = 2, 3, 4 where X and Y are different halogens and A (Aniline ring side) and B (Benzoyl ring side) represent the different phenyl rings] of mixed dihalogenated benzanilides; For example, 2FA–2IB [molecular formula = C13H9N1OFI] corresponds to 2-iodo-N-(2-fluorophenyl)benzamide. In the case of fluorine, at either the ortho or meta position on either the A or B side of the ring, it exhibits disorder in the crystal structure.](/image/article/2012/CE/c1ce05441d/c1ce05441d-s1.gif) |
| | Scheme 1 Schematic representation [nnXA–nnYB; n = 2, 3, 4 where X and Y are different halogens and A (Aniline ring side) and B (Benzoyl ring side) represent the different phenyl rings] of mixed dihalogenated benzanilides; For example, 2FA–2IB [molecular formula = C13H9N1OFI] corresponds to 2-iodo-N-(2-fluorophenyl)benzamide. In the case of fluorine, at either the ortho or meta position on either the A or B side of the ring, it exhibits disorder in the crystal structure. | |
Experimental
The dihalogenated benzanilides were prepared as followed in the literature.15 The purity of samples was characterized by IR, NMR and Powder X-ray data. Single crystals of desired size and quality were grown by slow evaporation at 0 °C and 25 °C by dissolving the substance in different solvent combinations such as ethyl acetate/hexane, ethyl acetate/acetonitrile and dichloromethane/hexane. Morphologies ranging from thin plates to blocks were obtained (see ESI†).
Crystal structure determination
Single crystals of required size were selected (Table 1) and some of these compounds were collected on a Bruker AXS SMART APEX CCD diffractometer with X-ray generator operating at 50 kV and 35 mA using graphite monochromated MoKα (λ = 0.7107 Å) radiation.16 The data for the rest of the compounds were recollected on CrysAlis CCD Xcalibur, Eos (Nova), Oxford Diffraction with X-ray generator operating at 50 kV and 1 mA, using MoKα radiation (λ = 0.7107 Å).17 Data were collected for 606 frames per set by using SMART with different settings of φ (0°, 90°, 180°) keeping the sample to detector distance of 6.062 cm and the 2θ value fixed at −25°. Data reduction and analysis were performed by SAINTPLUS, XPREP and SADABS.16 The structures were solved and refined by using SHELXL97,18 using the program suite WinGX.19 The molecular diagrams were generated using POV-Ray20 and the packing diagrams were generated using Mercury.21 The geometric calculations were carried out by PARST9522 and PLATON.23 All hydrogen atoms were positioned geometrically (C–H = 0.93 Å, N–H = 0.86 Å), and refined using a riding model with Uiso(H) = 1.2Ueq(C, N). The disorder was analyzed using the PART command in SHELXL-9718 and the disordered fluorine along with associated carbon atoms of the phenyl ring were treated and refined with two independent positions, namely A and B. For the purpose of refinement, the carbon positions for A and B are fixed at the same value. Table 1 lists all crystallographic parameters. Tables 2 and 3 list all intra-, intermolecular interactions and halogen⋯halogen interactions, respectively. The H⋯F distances considered for analysis of the supramolecular motifs are below 3.0 Å [the sum of the van der Waals radii + 0.4 Å24] and the corresponding angles considered are greater than or equal to 110°.7Table 4 lists the relevant C–F bond lengths for both the fluorine atoms (major and minor occupancy) exhibiting positional disorder. C–H⋯F–C interactions, wherein the occupancy of disordered fluorine atom is greater than or equal to 0.4, have only been considered for analysis of the crystal packing features.
Table 1 Crystallographic information
| |
2FA–
2IB
|
3FA–
2ClB
|
3FA–
2BrB
|
3FA–
2IB
|
3FA–
3BrB
|
| Formula |
C13H9NOFI
|
C13H9NOFCl
|
C13H9NOFBr
|
C13H9NOFI
|
C13H9NOFBr
|
| Formula weight |
341.1 |
249.7 |
294.1 |
341.1 |
294.1 |
| Temperature/K |
292(2) |
292(2) |
292(2) |
292(2) |
292(2) |
| Size of crystals |
0.22 × 0.18 × 0.13 |
0.30 × 0.19 × 0.11 |
0.24 × 0.23 × 0.16 |
0.2 × 0.14 × 0.10 |
0.25 × 0.19 × 0.14 |
| CCDC no. |
811042
|
764629
|
764625
|
774717
|
764626
|
| Space group |
Pna21 |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P
|
P212121 |
|
a/Å |
9.0447(2) |
13.647(3) |
5.090(1) |
5.101(3) |
4.371(1) |
|
b/Å |
14.1185(3) |
12.338(3) |
10.695(2) |
11.019(1) |
5.192(1) |
|
c/Å |
9.9862(3) |
13.731(3) |
12.054(2) |
12.441(1) |
50.481(4) |
|
α/° |
90 |
90 |
113.637(3) |
64.411(3) |
90 |
|
β/° |
90 |
90.686(2) |
97.827(3) |
79.243(3) |
90 |
|
γ/° |
90 |
90 |
93.133(25) |
87.180(3) |
90 |
| Volume/Å3 |
1275.2(1) |
2311.9(1) |
591.3(1) |
619.2(11) |
1145.6(2) |
|
Z
|
4 |
8 |
2 |
2 |
4 |
| Density/gcm−3 |
1.78 |
1.43 |
1.65 |
1.83 |
1.71 |
|
μ/mm−1 |
2.506 |
0.324 |
3.470 |
2.580 |
3.582 |
|
F (000) |
656.0 |
1024.0 |
292.0 |
328.0 |
584.0 |
|
θ
min, max/° |
2.5, 26.0 |
3.3, 26.0 |
1.9, 25.7 |
1.9, 25.0 |
1.6, 26.0 |
|
h
min, max
|
−10, 11 |
−16, 16 |
−6, 6 |
−6, 6 |
−5, 5 |
|
k
min, max
|
−17, 17 |
−15, 15 |
−13, 13 |
−13, 12 |
−6, 6 |
|
l
min, max
|
−11, 12 |
−16, 16 |
−14, 14 |
−14, 14 |
−62, 62 |
| No. of refln meas. |
7168 |
23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 929 929 |
6023 |
6993 |
11914 |
| No. unique refln |
2362 |
4531 |
2239 |
2143 |
2267 |
| No. of parameter |
164 |
327 |
164 |
165 |
161 |
|
R_all, R_obs |
0.044, 0.034 |
0.070, 0.045 |
0.039, 0.030 |
0.030, 0.025 |
0.042, 0.034 |
| wR2_all, wR2_obs |
0.071, 0.069 |
0.123, 0.129 |
0.069, 0.065 |
0.056, 0.054 |
0.069, 0.067 |
|
Δρ
max, min/e Å−3 |
0.463, −0.640 |
0.504, −0.338 |
0.238, −0.358 |
0.549, −0.483 |
0.331, −0.234 |
| GoF |
1.006 |
1.083 |
1.050 |
1.041 |
1.140 |
| |
2FB–
2ClA
|
2FB–
2BrA
|
2FB–2IA
|
3FB–
4ClA
|
3FB–
4BrA
|
| Formula |
C13H9NOFCl
|
C13H9NOFBr
|
C13H9NOFI
|
C13H9NOFCl
|
C13H9NOFBr
|
| Formula weight |
249.7 |
294.1 |
341.1 |
249.7 |
294.1 |
| Temperature/K |
292(2) |
292(2) |
292(2) |
292(2) |
292(2) |
| Size of crystals |
0.30 × 0.28 × 0.19 |
0.30 × 0.24 × 0.11 |
0.30 × 0.28 × 0.22 |
0.28 × 0.19 × 0.1 |
0.29 × 0.17 × 0.09 |
| CCDC no. |
764627
|
640039
|
774718
|
764628
|
640042
|
| Space group |
P21/n |
P21/n |
P21/n |
C2/c |
C2/c |
|
a/Å |
11.018(2) |
11.219(2) |
11.547(1) |
25.686(6) |
26.081(3) |
|
b/Å |
4.743(7) |
4.741(2) |
4.767(1) |
5.242(1) |
5.235(7) |
|
c/Å |
21.678(3) |
21.685(3) |
21.939(2) |
17.398(4) |
17.548(2) |
|
α/° |
90 |
90 |
90 |
90 |
90 |
|
β/° |
94.101(2) |
93.910(2) |
92.302(5) |
104.252(1) |
103.716(2) |
|
γ/° |
90 |
90 |
90 |
90 |
90 |
| Volume/Å3 |
1129.9(1) |
1150.7(1) |
1206.6(3) |
2270.8(1) |
2327.6(1) |
|
Z
|
4 |
4 |
4 |
8 |
8 |
| Density/gcm−3 |
1.47 |
1.70 |
1.88 |
1.46 |
1.68 |
|
μ/mm−1 |
0.332 |
3.566 |
2.648 |
0.330 |
3.526 |
|
F (000) |
511.9 |
584.0 |
655.9 |
1024.0 |
1168.0 |
|
θ
min, max/° |
1.9, 26.0 |
2.0, 25.7 |
1.9, 26.0 |
1.6, 25.5 |
1.6, 25.7 |
|
h
min, max
|
−13, 13 |
−13, 13 |
−14, 13 |
−30, 30 |
−31, 31 |
|
k
min, max
|
−5, 5 |
−5, 5 |
−5, 5 |
−5, 6 |
−6, 6 |
|
l
min, max
|
−26, 26 |
−26, 23 |
−27, 26 |
−21, 21 |
−21, 21 |
| No. of refln meas. |
10905 |
8146 |
24409 |
17504 |
8426 |
| No. unique refln |
2216 |
2179 |
2372 |
2112 |
2220 |
| No. of parameter |
164 |
152 |
164 |
164 |
164 |
|
R_all, R_obs |
0.053, 0.041 |
0.041, 0.031 |
0.030, 0.024 |
0.059, 0.040 |
0.048, 0.034 |
| wR2_all, wR2_obs |
0.104, 0.098 |
0.072, 0.070 |
0.049, 0.047 |
0.153, 0.125 |
0.085, 0.077 |
|
Δρ
max, min/e Å−3 |
0.219, −0.177 |
0.576, −0.265 |
0.423, −0.505 |
0.234, −0.207 |
0.415, −0.326 |
| GoF |
1.063 |
1.055 |
1.043 |
1.124 |
1.027 |
Table 2 Intra- and intermolecular interactionsa
| |
D–H⋯A |
D–H/Å |
D⋯A/Å |
X⋯A/Å |
∠D–X⋯A/° |
Symmetry |
|
X = H or halogen; Cg1 = centroid of the ring C1–C6, Cg2 = C8–C13, Cg3 = C21–C26, Cg4 = C28–C33.
|
|
2FA–2IB
|
N1–H1⋯O1 |
0.86 |
2.857(5) |
2.04 |
157 |
1/2 + x, 1/2 − y, z |
| C4–H4⋯F1A |
0.93 |
3.140(7) |
2.60 |
117 |
−x, −y + 1, +z + 1/2 |
| C4–H4⋯F1B |
0.93 |
3.351(8) |
2.58 |
141 |
−x − 1/2, +y − 1/2, +z + 1/2 |
| C2–H2⋯Cg2 |
0.93 |
3.593(4) |
2.81 |
142 |
1/2 + x, 1/2 − y, z |
|
3FA–
2ClB
|
C13–H13⋯O1 |
0.93 |
2.882(2) |
2.34 |
117 |
x, y, z |
| C33–H33⋯O2 |
0.93 |
2.913(3) |
2.45 |
111 |
x, y, z |
| N1–H1⋯O2 |
0.86 |
2.977(2) |
2.13 |
170 |
1/2 + x, 1/2 − y, −1/2 + z |
| N2–H2⋯O1 |
0.86 |
2.877(2) |
2.04 |
164 |
1/2 + x, 1/2 − y, −1/2 + z |
|
Cg2 ⋯Cg4 |
— |
3.953(2) |
— |
— |
1 − x, −y, 1 − z |
|
3FA–
2BrB
|
N1–H1⋯O1 |
0.86 |
2.930(3) |
2.18 |
146 |
−1 + x, y, z |
| C13–H13⋯O1 |
0.93 |
2.893(3) |
2.40 |
113 |
x, y, z |
| C3–H3⋯Cg1/C13 |
0.93 |
3.693(2) |
3.04/2.88 |
146 |
−x + 1, −y, −z + 2 |
| C6–Br1⋯Cg2/C2 |
— |
5.293(1) |
4.10/3.49 |
154 |
−x, −y + 1, −z + 2 |
|
3FA–2IB
|
N1–H1⋯O1 |
0.86 |
2.948(3) |
2.18 |
148 |
1 + x, y, z |
| C13–H13⋯O1 |
0.93 |
2.908(4) |
2.42 |
113 |
x, y, z |
| C3–H3⋯Cg1/C13 |
0.93 |
3.703(2) |
3.26/2.88 |
162 |
−x + 1, −y, −z + 2 |
| C6–I1⋯Cg2/C2 |
— |
5.708(2) |
4.10/3.619 |
174 |
−1 + x, y, z |
|
3FA–
3BrB
|
N1–H1⋯O1 |
0.86 |
3.031(3) |
2.21 |
159 |
x, 1 + y, z |
| C13–H13⋯O1 |
0.93 |
2.944(4) |
2.53 |
108 |
x, y, z |
|
2FB–
2ClA
|
C13–H13⋯O1 |
0.93 |
2.896(3) |
2.43 |
111 |
x, y, z |
| N1–H1⋯F1A |
0.86 |
2.766(2) |
2.23 |
120 |
x, y, z |
| N1–H1⋯O1 |
0.86 |
2.932(2) |
2.22 |
140 |
x, 1 + y, z |
| C12–H12⋯O1 |
0.93 |
3.422(3) |
2.64 |
142 |
−x + 1, −y + 1, −z |
| C9–Cl1⋯Cg2/C4 |
— |
4.019(2) |
3.72/3.47 |
166 |
x, 1 + y, z |
|
2FB–
2BrA
|
C13–H13⋯O1 |
0.93 |
2.894(3) |
2.44 |
110 |
x, y, z |
| N1–H1⋯F1A |
0.86 |
2.784(3) |
2.27 |
118 |
x, y, z |
| N1–H1⋯O1 |
0.86 |
2.926(3) |
2.20 |
143 |
x, 1 + y, z |
| C12–H12⋯O1 |
0.93 |
3.427(4) |
2.63 |
143 |
−x, −y − 1, −z |
| C9–Br1⋯Cg2/C4 |
— |
4.025(3) |
3.74/3.48 |
168 |
x, 1 + y, z |
|
2FB–2IA
|
C13–H13⋯O1 |
0.93 |
2.919(4) |
2.50 |
108 |
x, y, z |
| N1–H1⋯F1A |
0.86 |
2.828(4) |
2.36 |
115 |
x, y, z |
| N1–H1⋯O1 |
0.86 |
2.895(3) |
2.13 |
148 |
x, 1 + y, z |
| C12–H12⋯O1 |
0.93 |
3.464(4) |
2.67 |
143 |
−x, −y − 1, −z |
| C9–I1⋯Cg2/C4 |
— |
4.063(3) |
3.88/3.64 |
169 |
x, 1 + y, z |
| |
D–H⋯A |
D–H/Å |
D⋯A/Å |
H⋯A/Å |
∠D–H⋯A/° |
Symmetry |
|
3FB–
4ClA
|
C13–H13⋯O1 |
0.93 |
2.920(3) |
2.46 |
111 |
x, y, z |
| N1–H1⋯O1 |
0.86 |
3.051(2) |
2.25 |
156 |
x, −1 + y, z |
| C9–H9⋯O1 |
0.93 |
3.440(1) |
2.62 |
127 |
−x + 1, −y − 1, −z |
| C6–H6⋯Cg2 |
0.93 |
3.621(3) |
2.87 |
138 |
1/2 − x, 1/2 − y, −z |
|
3FB–
4BrA
|
C13–H13⋯O1 |
0.93 |
2.923(4) |
2.47 |
110 |
x, y, z |
| N1–H1⋯O1 |
0.86 |
3.042(3) |
2.24 |
154 |
x, −1 + y, z |
| C9–H9⋯O1 |
0.93 |
3.243(4) |
2.59 |
127 |
x, −1 + y, z |
| C6–H6⋯Cg2 |
0.93 |
3.624(3) |
2.88 |
138 |
1/2 − x, 3/2 − y, −z |
| C13–H13⋯Cg2 |
0.93 |
3.549(3) |
2.89 |
129 |
1/2 − x, 1/2 + y, 1/2 − z |
Table 3
Halogen⋯halogen interactions
| Compound |
C–X⋯Y–C |
X⋯Y/Å |
∠C–X⋯Y/° |
∠C–Y⋯X/° |
Symmetry |
Type |
|
3FA–
2BrB
|
C6–Br1⋯F1A–C10 |
3.193(7) |
90.9 |
165.8 |
−x + 1, −y + 1, −z + 2 |
II |
|
3FA–2IB
|
C6–I1⋯F1A–C10 |
3.334(3) |
93.7 |
158.5 |
−x + 1, −y + 2, −z + 1 |
II |
|
3FA–
3BrB
|
C5–Br1⋯Br1–C5 |
3.686(7) |
101.8 |
168.5 |
−x + 2, y − 1/2, −z + 3/2 |
II |
|
3FB–
4ClA
|
C3–F1A⋯Cl1–C11 |
3.119(8) |
116.6 |
172.0 |
x + 1/2, −y + 1/2, z + 1/2 |
II |
| C5–F1B⋯Cl1–C11 |
3.174(4) |
79.6 |
132.6 |
x + 1/2, −y + 3/2, −z + 1 |
— |
|
3FB–
4BrA
|
C3–F1A⋯Br1–C11 |
3.185(3) |
172.2 |
118.3 |
x + 1/2, −y + 3/2, z + 1/2 |
II |
| C5–F1B⋯Br1–C11 |
3.227(4) |
77.4 |
132.9 |
−x + 1/2, −y + 3/2, −z + 2 |
— |
Table 4 C–F bond lengths, involving disordered fluorine, as a function of their major occupancy
| |
Major C–F bond Length/Å |
Minor C–F bond Length/Å |
Major occupancy fluorine (F1) in % |
|
Two molecules in the asymmetric unit and both are exhibiting disorder in fluorine.
|
|
2FA–2IB
|
1.288 |
1.237 |
58 |
|
3FA–
2ClB
a
|
1.336/1.342 |
1.091/1.181 |
79/92 |
|
3FA–
2BrB
|
1.312 |
1.159 |
73 |
|
3FA–2IB
|
1.319 |
1.108 |
81 |
|
3FA–
3BrB
|
1.319 |
1.273 |
67 |
|
2FB–
2ClA
|
1.351 |
1.313 |
88 |
|
2FB–
2BrA
|
1.345 |
1.314 |
83 |
|
2FB–2IA
|
1.311 |
1.320 |
62 |
|
3FB–
4ClA
|
1.324 |
1.317 |
52 |
|
3FB–
4BrA
|
1.324 |
1.293 |
61 |
|
Ref. 6
|
1.337 |
1.289 |
67 |
| 1.327 |
1.148 |
70 |
| 1.296 |
1.262 |
63 |
|
Ref. 8
|
1.326 |
1.159 |
86 |
| 1.306 |
1.296 |
60 |
| 1.313 |
1.283 |
62 |
|
Ref. 9
|
1.255 |
1.232 |
53 |
| 1.320 |
1.132 |
79 |
| 1.340 |
1.155 |
89 |
|
Ref. 10
|
1.204 |
— |
50 |
| 1.310 |
1.175 |
73 |
|
Ref. 13
|
1.348 |
1.130 |
85 |
Results and discussions
2FA–2IB (2-iodo-N-(2-fluorophenyl)benzamide, C13H9NOFI) crystallizes in the non-centrosymmetric orthorhombic Pna21 space group (Z = 4) (Table 1). This shows positional disorder at the fluorine atom (Fig. 1). The value of the major occupied fluorine (F1) atom is 0.58(2). The N–H⋯O hydrogen bond forms an infinite chain along the a-axis. The major and minor occupied fluorine atoms prefer bifurcated weak C–H⋯F hydrogen bonds (utilizing the nglide and 21 screw axis as key symmetry elements) with additional C–H⋯π interactions stabilizing the zig-zag molecular packing (Fig. 2 and Table 2). However, in the case of 2FA–2FB, 2FA–2ClB and 2FA–2BrB, i.e. on replacement of the iodine atom in 2FA–2IB with fluorine, chlorine and bromine respectively, positional disorder at the fluorine position is eliminated.5 The molecular assembly of 2FA–2IB is very different from 2FA–2FB, 2FA–2ClB and 2FA–2BrB. It is of interest to compare the molecular conformation in all these four compounds containing different halogens. On going from fluorine to chlorine to bromine to iodine the dihedral angles between the two phenyl rings are found to be 1, 6, 72, 87°, respectively. The resulting equilibrium conformation is found to result from a minimization of the C–F/C–Cl/C–Br/C–I/CO dipolar interactions and maximization of the intramolecular interactions depending on the halogen present. It is noteworthy that this value is the least in the case of fluorine, again pointing to the significance of N–H⋯F interactions in stabilizing the molecular geometry.
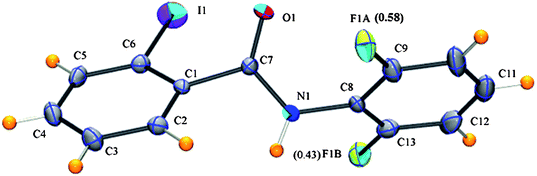 |
| | Fig. 1
ORTEP diagram of 2FA–2IB drawn with 50% ellipsoidal probability, highlighting the atom-numbering scheme, along with the occupancies for the disordered fluorine. The disordered hydrogen and carbon atoms have been omitted for clarity. | |
 |
| | Fig. 2 The infinite N–H⋯O hydrogen bonds along the a-axis with the C–H⋯F hydrogen bond stabilizing the zig-zag molecular packing in 2FA–2IB. | |
3FA–
2ClB, (2-chloro-N-(3-fluorophenyl)benzamide, C13H9NOFCl) crystallizes in the centrosymmetric monoclinic space groupP21/n with Z′ = 2 (Z = 8) (Table 1). Both symmetry independent molecules show positional disorder at the meta site of the aniline ring with different occupancies [F1A = 0.79(1), F1B = 0.21(1) and F2A = 0.92(2), F2B = 0.08(2)]. The discrepancy in the occupancy of disordered fluorine atom and the variation in their torsion around the amide bond [C7–C1–N1–C8 = 171.5(2) and C27–C21–N2–C28 = 177.7(1)°] serve as the key differences between the two independent molecules. As is common in all substituted benzanilides, the N–H group is trans to the CO group resulting in the formation of a C–H⋯O intra-molecular interaction (Fig. 3).
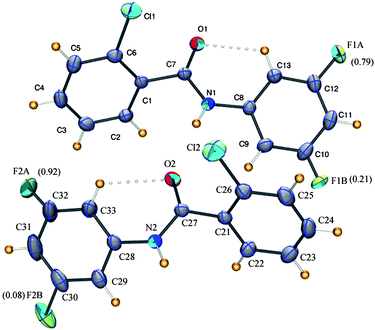 |
| | Fig. 3
ORTEP diagram shows two molecules in the asymmetric unit of 3FA–2ClB along with the occupancies for the disordered fluorine. Intra-molecular interactions are joined by open circles. The disordered hydrogen and carbon atoms have been omitted for clarity. | |
Well defined N–H⋯O hydrogen bonds, as seen in earlier studied benzanilides, stabilize the packing of the molecule in the crystal lattice by generating a chain running diagonal to the ac plane. An additional π⋯π interaction develops to hold the two symmetry independent molecules (Fig. 4). The variation in C–F bond lengths associated with the major and minor occupied fluorine atoms is depicted in Table 4. The table also highlights the C–F bond lengths in molecules containing disordered organic fluorine in the molecular structure.6,8–10,12
 |
| | Fig. 4 The infinite chains through N–H⋯O hydrogen bonds whereas π⋯π acts as a link between the chains in 3FA–2ClB. | |
3FA–
2BrB, 2-bromo-N-(3-fluorophenyl)benzamide (C13H9NOFBr) crystallizes in the centrosymmetric triclinic P space group with Z = 2 (Table 1). The corresponding iodine molecule 3FA–2IB (2-iodo-N-(3-fluorophenyl)benzamide, C13H9NOFI) is isostructural and both show positional disorder at the fluorine atom (Fig. 5). The values of major occupied fluorine (F1A) atoms are 0.73(2) and 0.81(1) for 3FA–2BrB and 3FA–2IB, respectively. As in the previous examples, the strong N–H⋯O hydrogen bond forms an infinite chain along the a-axis. In addition, C–H⋯π along with C–X⋯π (X = Br or I) interaction generates a new feature in the crystal packing resulting in a stair case geometry as shown in Fig. 6.
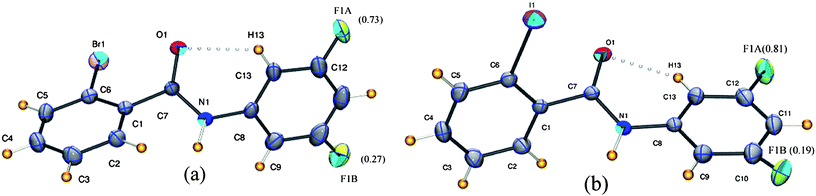 |
| | Fig. 5
ORTEP diagram of 3FA–2BrB (a) and 3FA–2IB (b) with the occupancies for the disordered fluorine. Intra-molecular interactions are joined by open circles. The disordered hydrogen and carbon atoms have been omitted for clarity. | |
 |
| | Fig. 6 Staircase geometry through Br⋯π and C–H⋯π dimer interaction in 3FA–2BrB. | |
On comparison of the molecular geometry of 3FA–2FB, 3FA–2ClB, 3FA–2BrB, 3FA–2IB, it is observed that the dihedral angles between the phenyl rings on going across the halogens fluorine to iodine are 1.5°, 53.2°, 54.8°, and 56.1°. In the case of 3FA–2FB, the molecular conformation is locked conformationally because of the presence of intramolecular N–H⋯F and C–H⋯O interactions. Thus although the fluorine atom is present in the meta position it exhibits no positional disorder. In the remaining cases, the dihedral angle between the B-ring with the plane containing the amido functionality increases from 27.3°, 36.2°, 83.6°, and 84.9° in the case of four halogens, fluorine to iodine, again signifying the importance of N–H⋯F in 3FA–2FB and C–Halogen⋯π interaction in 3FA–2BrB and 3FA–2IB respectively. In all the four compounds, the conformation between the A-ring and amido group varies between 17.0° and 28.6° pointing to the fact that C–H⋯O intramolecular interaction is conserved in all the molecular geometries. One point of significance is that the compound 3FA–2FB crystallizes in the non-centrosymmetric space group Pca21, whereas the corresponding chloro, bromo and iodo derivatives crystallize in the centrosymmetric space group, again pointing to the significance of intermolecular C–H⋯F hydrogen bonds which dictate crystal packing in the difluorinated compound.7
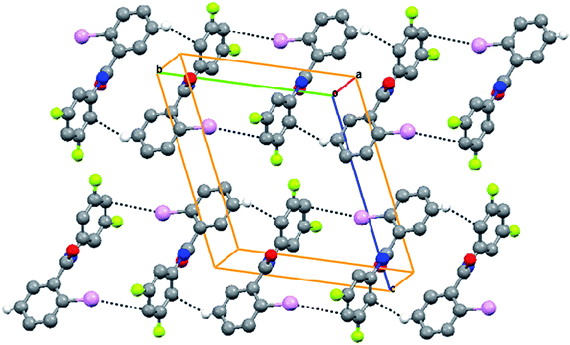
3FA–
3BrB (3-bromo-N-(3-fluorophenyl)benzamide, C13H9NOFBr) crystallizes in the non-centrosymmetric orthorhombic P212121 space group with fluorine disorder at both possible meta sites of the aniline ring. The fluorine atom F1A has a major occupancy of 0.67(1). The N–H bond is trans to the CO group (Fig. 7). The crystal packing is mainly through infinite N–H⋯O hydrogen bond chains along the b-axis. Further, homo halogen⋯halogen (Br⋯Br) Type II contacts form a chain running parallel to the N–H⋯O chain (Fig. 8, Tables 2 and 3). The corresponding chloro and iodo derivatives have not been structurally characterized, although the corresponding 3FA–3FB compound has been observed to crystallize as concomitant dimorphs wherein both the forms exhibit positional disorder in both the fluorine atoms present in the meta position on both the phenyl rings.8
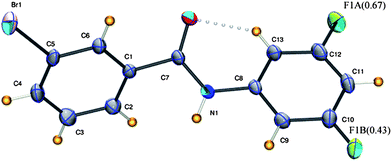 |
| | Fig. 7
ORTEP diagram of 3FA–3BrB with the occupancies for the disordered fluorine. Intra-molecular interaction is joined by open circles. The disordered hydrogen and carbon atoms have been omitted for clarity. | |
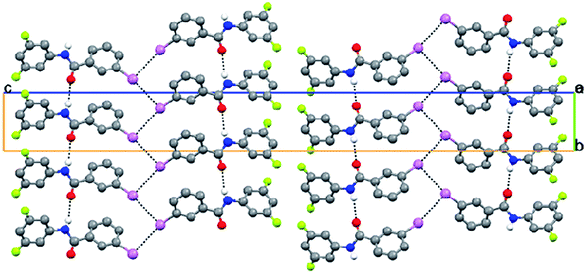 |
| | Fig. 8 N–H⋯O infinite chains are interconnected through Br⋯Br short contacts in 3FA–3BrB. | |
2FB–
2ClA (2-fluoro-N-(2-chlorophenyl)benzamide, C13H9NOFCl), 2FB–2BrA (2-fluoro-N-(2-bromophenyl)benzamide, C13H9NOFBr), and 2FB–2IA(2-fluoro-N-(2-iodophenyl)benzamide, C13H9NOFI) are isostructural and crystallize in the centrosymmetric monoclinic P21/n space group with Z = 4 molecules in the unit cell. The ortho site fluorine atom shows positional disorder with major occupancy (F1A) providing the rigid N–H⋯F intra-molecular hydrogen bond towards molecular conformation (Fig. 9). This feature once again proves the rigidity provided by the N–H⋯F intra-molecular interaction. It is of interest to note that the occupancy at F1A reduces from 0.88(2) in 2FB–2ClA to 0.83(1) in 2FB–2BrA and 0.62(2) in the case of 2FB–2IA. The N–H⋯O hydrogen bonds generate a chain along the b-axis. Additional C–H⋯O interactions stabilize a herringbone type molecular geometry as shown in Fig. 10 (Table 2). In all three cases, additional C–X⋯π (X = Cl, Br, I) interaction provide further stability (Fig. 10 and Table 2).
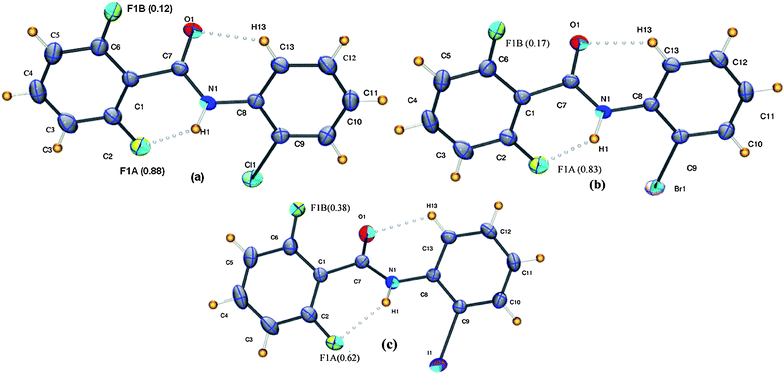 |
| | Fig. 9
ORTEP diagram of 2FB–2ClA, 2FB–2BrA and 2FB–2IA with the occupancies for the disordered fluorine. Intra-molecular interactions are joined by open circles. The disordered hydrogen and carbon atoms have been omitted for clarity. | |
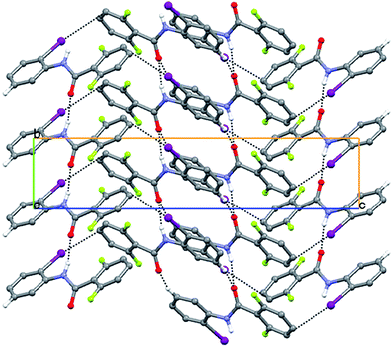 |
| | Fig. 10 The herringbone type packing through N–H⋯O hydrogen bonds of infinite chains linked through, C–X⋯π (where X = Cl/Br/I) interaction for 2FB–2ClA, 2FB–2BrA and 2FB–2IA, with additional C–H⋯O interactions. | |
It is interesting to note that the molecular conformation of all the three molecules, namely 2FB–2ClA, 2FB–2BrA, and 2FB–2IA, exhibit similarity in the dihedral angles [Table S1†]. It is to be noted that the C–F dipole is always oriented away from the CO dipole to minimize dipole–dipole repulsions in the solid state and facilitate the formation of intramolecular N–H⋯F and C–H⋯O intramolecular interactions. The molecular conformation is analogous to the one observed in the triclinic form of benzanilide.25 Furthermore, this indicates that the presence of varying halogens, namely chloro, bromo and iodo atoms, on the ortho position presents invariant electronic features associated with these molecules.
3FB–
4ClA, (3-fluoro-N-(4-chlorophenyl)benzamide, C13H9NOFCl) and 3FB–4BrA, (3-fluoro-N-(4-bromophenyl)benzamide, C13H9NOFBr) are isostructural and crystallize in the centrosymmetric monoclinic C2/c space group with Z = 8. Both the structures show the usual intra- and intermolecular interactions and disorder in the fluorine atom is nearly 0.52(2) in the chlorine (3FB–4ClA) structure while it is 0.61(2) in the bromine (3FB–4BrA) structure (Fig. 11). N–H⋯O hydrogen bonds generate chains along the b-axis which are interconnected through hetero Br⋯F/Cl⋯F (Type II) contacts which involve the fluorine atom with major occupancy. Additional C–H⋯O and C–H⋯π intermolecular interactions assist the molecular assembly in the crystal.
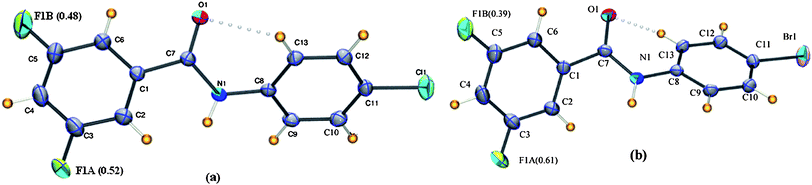 |
| | Fig. 11
ORTEP diagram of (a) 3FB–4ClA and (b) 3FB–4BrA with the occupancies for the disordered fluorine. Intra-molecular interactions are joined by open circles. The disordered hydrogen and carbon atoms have been omitted for clarity. | |
It is of interest to compare the molecular structures of 3FB–4ClA/4BrA with related benzanilides. It is observed that the space group of these compounds is similar to 3-chloro-N-(4-methylphenyl)benzamide[ClMe] and the monoclinic form of benzanilide26 wherein all the compounds crystallize in the centrosymmetric C2/c space group. A related compound (3-fluoro-N-(4-methylphenyl)benzamide)[FMe] crystallizes in the P21/c space group, the relationship with the previous compounds being a doubling of the c-axis. It is noteworthy that the molecular conformation of 3FB–4ClA/4BrA is similar to ClMe/FMe along with the monoclinic and triclinic forms of benzanilide. Furthermore, the unit cell volume for ClMe is greater than both 3FB–4ClA/4BrA. This indicates that hetero Br⋯F/Cl⋯F halogen contacts are indeed attractive in nature. Experimental and theoretical charge density studies27 performed on 2-chloro-4-fluorobenzoic acid bring out the attractive nature associated with Cl⋯F interactions in the solid state (Fig. 12).
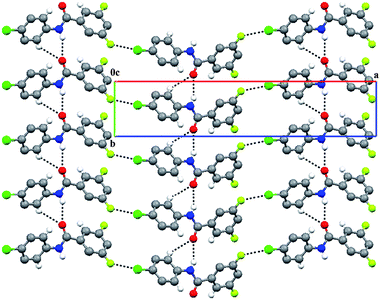 |
| | Fig. 12 N–H⋯O infinite chains are linked through Cl/Br⋯F short contacts in the case of 3FB–4ClA and 3FB–4BrA. | |
There are many related compounds, namely 4-fluoro-N-(3-fluorophenyl)benzamide and 4-fluoro-N-(2-fluorophenyl)benzamide, which exhibit similarity in molecular geometry. The latter crystallizes as dimorphs, having a fluorine atom in the meta position, but does not exhibit any positional disorder related to fluorine. This is because the formation of the molecular and crystal structure is a delicate balance of the factors which involve minimizing inter-electronic repulsions in different portions of the molecule and maximization of the intermolecular interactions. The existence of crystal disorder is advantageous because the molecule may have an entropic or enthalpic gain, or both, in terms of different independent conformations with increase in the number of intermolecular contacts which can be attractive/repulsive. If disorder is absent it signifies that the energetic gain from disorder is less than when the molecule is ordered, and the stability in terms of molecular conformation (minimum dipolar repulsions) becomes more important and contributes towards the stability of crystal packing in terms of the subsequent intermolecular interactions which develop in the crystal. Such is the case in the above-mentioned two compounds.
It is also observed that disorder can prevail, yet there is no enthalpic gain in terms of increased intermolecular interactions involving disorder of the fluorine atom when present in the meta position. Such is the case in the unsolvated and solvated forms of 3-fluorobenzoylaminophenyl-3-fluorobenzoate, wherein the conformation of the benzoate part remains the same, and although the fluorine present on this ring exhibits disorder, due to C–C bond rotation of the fluorophenyl ring, in the solvated form and none in the unsolvated form, there are no intermolecular contacts involving these.
Cambridge structural database (CSD) analysis
The CSD (Version 5.32, 2010) Database analysis28 was performed to get insights into the disordered nature of halogens substituted at the ortho or meta position on the phenyl ring of a compound. The search was restricted to compounds which have less than 10% R-factor, no free ions, only organic compounds and not polymeric. Table 5 is a representation of the number of structures which are listed as disordered. For example, the second column represented by 2-F refers to compounds which have orthofluorine substitution. The rows show the total number of structures followed by those which are ordered and those which are disordered. The last row gives the overall percentage of disordered structures. It is of interest to note that fluorine has the highest percentage of disordered structures. Fig. 13 shows a very interesting trend. The disorder is confined mostly to the fluorine atom at either the ortho or meta position. In contrast, Cl, Br and I not only show lower percentage of disorder but also have a negligible number of structures where disorder is localized at the halogen atom.
Table 5
CSD analysis results for ortho and meta halogenated compounds
| Ortho/meta number of hits |
2–F |
3–F |
2–Cl |
3–Cl |
2Br
|
3–Br |
2–I |
3–I |
| Total structures |
2869 |
2830 |
3631 |
3994 |
1447 |
1988 |
651 |
552 |
| Ordered structures |
2334 |
2313 |
3183 |
3516 |
1271 |
1749 |
558 |
474 |
| Disordered structures |
535 |
512 |
448 |
457 |
174 |
232 |
93 |
66 |
| Percentage (%) of disorder |
18.6 |
18.0 |
12.3 |
11.4 |
12.0 |
11.6 |
14.2 |
11.9 |
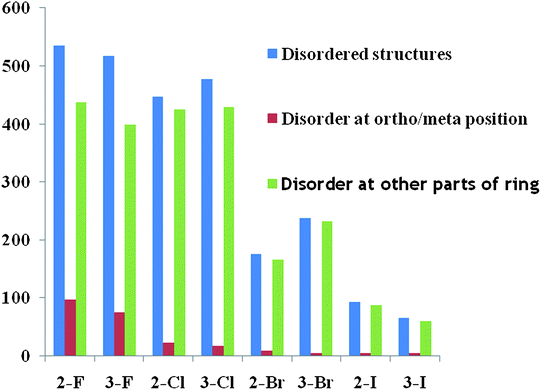 |
| | Fig. 13 Histogram representing the nature of disorder associated with a given halogen atom. | |
It is to be noted from the CSD search performed with the above-mentioned restrictions that the crystal disorder present in different molecules with varying halogen substitution brings out the significance of the fluorine atom and the fact that its participation in the formation of disordered structures is the highest. On increasing the size of the halogen, its involvement in getting statistically disordered over different sites is drastically reduced. This is associated with the build up of crystal disorder in different portions of the molecule. Initially, one might infer that the size of H and F being comparable to each other allows fluorine to get disordered. This can be a limiting factor for the heavier halogens and hence the associated disorder is reduced. But replacement of H by F, if it were purely the size effect, must result in isostructurality (and if possible isomorphism) in the solid state. But it has now been established that such a substitution results in altered packing modes in crystalline solids3 (sometimes the space group changes from centrosymmetric to non-centrosymmetric and vice versa) and this brings out the importance of weak intermolecular C–H⋯F interactions (now termed as “weak hydrogen bond”) in dictating molecules to pack in solids. Even if isostructurality prevails, the possibility of formation of such interactions cannot be ruled out. In this regard, it may be mentioned that disorder in the position of organic fluorine in molecules alters the crystal structure so as to facilitate the formation of weak C–H⋯F hydrogen bond which stabilizes the molecular packing.
It is also to be observed from a detailed analysis of the disorder present in the heavier halogenated molecules (differing ortho and meta position) [Table S2†] that related molecules have disorder associated with solvent molecules, or part or full disorder associated with the actual molecule. Thus, in the absence of heavy halogen disorder, the associated crystal structures achieve stability by allowing disorder to prevail in different portions of the molecule leading to an entropic gain. As this disorder involves fragments (composed of many atoms) the overall gain due to configurational entropy is substantial and leads to overall stability.
Table S2† [obtained from CSD] brings out the salient features associated with disorder between chemically different functional groups having comparable sizes which can get disordered over different positions in the molecule. It is noteworthy that fluorine prefers to exchange with hydrogen only because of comparable sizes. However, chlorine undergoes exchange thereby exhibiting positional disorder with the bromine [REFCODE: FOMLOJ & SEDDUA], nitro [JOCMIY, RAZZOI & XIKKOS] and methyl groups [IHATIU, LATWIN & NULVOG]. Bromine apart from exhibiting positional disorder with chlorine, methyl and nitro groups also shows positional disorder with the methoxy group [EBOBEC] and iodo atom [AJISAO].
Conclusions
The analysis of positional disorder associated with halogen substitution at either the ortho or meta position brings out several interesting features in crystal packing. Other significant observations are: (i) Fluorine substitution displays a high percentage of disorder. (ii) Disorder is confined to fluorine positions only. However, for heavier halogens, it is found at other atom sites. (iii) Fluorine involved interactions are maximized in disordered systems (because of fluorine disorder) wherein the same is compensated in heavier halogensvia disorder in different parts of the molecule.
Acknowledgements
We thank DST, India for the XRD facility at IISc and SKN thanks CSIR & DST for financial assistance. DC thanks DST for funding under the Fast Track Scheme. TNG thanks DST for the award of the J. C. Bose fellowship.
References
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
-
(a) G. Valero, X. Companyo and R. Rios, Chem.–Eur. J., 2011, 17, 2018–2037 CrossRef CAS;
(b) N. H. Campbell, D. L. Smith, A. P. Reszka, S. Neidle and D. O'Hagan, Org. Biomol. Chem., 2011, 9, 1328–1331 RSC.
- D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 2175–2186 RSC.
-
(a) M. Y. Anzahaee, J. K. Watts, N. R. Alla, A. W. Nicholson and M. J. Damha, J. Am. Chem. Soc., 2011, 133, 728–731 CrossRef CAS;
(b) D. Chopra, K. Nagarajan and T. N. Guru Row, Cryst. Growth Des., 2005, 5, 1035–1039 CrossRef CAS.
- S. K. Nayak, M. K. Reddy, T. N. Guru Row and D. Chopra, Cryst. Growth Des., 2011, 11, 1578–1596 CAS.
-
(a) A. R. Choudury and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 47–52 CrossRef;
(b) A. R. Choudury and T. N. Guru Row, CrystEngComm, 2006, 8, 265–274 RSC.
- D. Chopra and T. N. Guru Row, CrystEngComm, 2008, 10, 54–67 RSC.
- D. Chopra and T. N. Guru Row, Cryst. Growth Des., 2008, 8, 848–853 CAS.
- D. Chopra and T. N. Guru Row, Cryst. Growth Des., 2006, 6, 1267–1270 CAS.
- D. Chopra, V. Thiruvenkatam and T. N. Guru Row, Cryst. Growth Des., 2006, 6, 843–845 CAS.
- L. Ming, G. W. Si, W. L. Rong, L. Y. Feng and Y. H. Zheng, J. Mol. Catal. A: Chem., 2006, 258, 133–138 CrossRef.
- S. K. Nayak, K. N. Venugopala, D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 591–605 RSC.
- Rajnikant, D. Watkin and G. Tranter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1452–1454 CrossRef.
- K. Ejsmont, A. A. Domański, J. B. Kyzioł and J. Zaleski, J. Mol. Struct., 2005, 753, 92–98 CrossRef CAS.
- D. Chopra and T. N. Guru Row, J. Mol. Struct., 2005, 733, 133–141 CrossRef CAS.
-
(a) (a) SMART (V 5.628), SAINT (V 6.45a), Sadbs, XPREP, SHELXTL, Bruker AXS Inc, Madison, WI, 2004 Search PubMed.
-
Oxford Diffraction, CrystAlis CCD and CrystAlis RED, Version 1.171.33.31, Oxford Difraction Ltd, Abingdon, Oxfordshire, England, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- L. J. Farrugia, WinGX (V 1.70.01), J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
-
Persistence of Vision Team, POV-RAY. Persistence of Vision, Raytracer Pty. Ltd, Victoria, Australia, 2004, http://www.povray.org/ Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS
http://www.ccdc.cam.ac.uk/mercury
.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 569 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed.
- I. Dance, New J. Chem., 2003, 27, 22–27 RSC.
- K. F. Bowes, C. Glidewell, J. N. Low, J. M. S. Skakle and J. K. Wardell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o1–o3 Search PubMed.
- S. Kashino, K. Ito and M. Haisa, Bull. Chem. Soc. Jpn., 1979, 52, 365–369 CrossRef CAS.
- V. R. Hathwar and T. N. Guru Row, Cryst. Growth Des., 2011, 11, 1338–1346 CAS.
- F. H. Allen, CSD (V5.32, 2010), Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.