Development of EPA Method 525.3 for the analysis of semivolatiles in drinking water†
Paul E. Grimmett* and Jean W. Munch
U.S. Environmental Protection Agency, Office of Research and Development, National Exposure Research Laboratory, 26 W. Martin Luther King Dr Cincinnati, OH 45268, USA. E-mail: grimmett.paul@epa.gov
First published on 9th October 2012
Abstract
The United States Environmental Protection Agency's (EPA) Office of Ground Water and Drinking Water (OGWDW) collects nationwide occurrence data on contaminants in drinking water using the Unregulated Contaminant Monitoring Regulations (UCMRs). The unregulated contaminants, which are potential candidates for future regulation, may be selected from the Drinking Water Contaminant Candidate List (CCL), or may be emerging contaminants, with the potential for inclusion on future CCLs. In October 2009, OGWDW published the third Drinking Water CCL (CCL 3). Not all of the chemicals on CCL 3 are included in existing analytical methods with sufficient sensitivity and specificity for UCMR monitoring. Some of the chemicals that require method development research fall into the category known as semi-volatile organic chemicals (SVOCs). Currently, EPA Method 525.2 is used to measure SVOCs for both drinking water compliance and UCMR monitoring. The method utilizes C-18 solid-phase extraction (SPE) for contaminant isolation and concentration, followed by full scan gas chromatography-mass spectrometry (GC-MS) detection. A group of 27 contaminants from CCL 3, a list of chemicals from EPA's National Homeland Security Research Center (NHSRC), as well as other emerging compounds of interest, were evaluated for inclusion into a new revision of the method, EPA Method 525.3. Method improvements include: (1) utilizing a polymeric-based SPE sorbent for greater retention across sample pH range, (2) enhanced method sensitivity using GC-MS with selected ion monitoring (SIM), (3) safer and more user-friendly sample preservatives, and (4) sample preparation techniques that help overcome matrix-induced chromatographic response enhancement. To be used for nationwide monitoring, the new method must be rugged across a range of drinking water sources, sensitive and highly specific to minimize false positives. It must also be cost effective and simple enough for commercial lab settings. Accuracy and precision data using full scan GC-MS mode are presented for 125 contaminants. Multi-laboratory data obtained in selected ion mode (SIM) are presented for a subset of priority analytes. Lowest Concentration Minimum Reporting Limits (LCMRLs) of priority analytes obtained from four separate laboratories ranged from 0.0006 to 0.28 μg L−1, with an average LCMRL of 0.035 μg L−1 and median LCMRL of 0.011 μg L−1, under SIM conditions.
1. Introduction
The 1996 amendments to the Safe Drinking Water Act (SDWA) require the United States Environmental Protection Agency (EPA) to establish a Drinking Water Contaminant Candidate List (CCL) of chemicals and microbes that the EPA will consider for future regulation. The first CCL was published in 1998, with updated versions scheduled every 5 years. The CCL 3 was published in October 2009 and contains 96 organic chemicals or chemical groups.1 For EPA to make a regulatory determination on a contaminant, nationwide occurrence data are necessary. The mechanism that EPA has established to collect large amounts of nationally based occurrence data is through Unregulated Contaminant Monitoring Regulations (UCMRs). In order for a contaminant to be included for monitoring in a UCMR, a standardized method for its measurement in drinking water must be available. Of the chemicals or chemical groups listed on CCL 3, over half are already in EPA drinking water methods or contained in methods currently being developed by EPA. Approximately 30% of the chemicals on the CCL 3 fall into the chemical category known as semi-volatile organic chemicals (SVOCs). While some of the listed SVOCs are included in existing methods with sufficient sensitivity and specificity for UCMR monitoring, others require method development research. EPA Method 525.2 (ref. 2) is a broad scan method for measuring SVOCs that is already in use for drinking water monitoring, both for contaminants already regulated under the SDWA (Table 1) and for emerging contaminants of concern under previous UCMRs. The method employs solid-phase extraction (SPE) and concentration, then analysis by gas chromatography-mass spectrometry (GC-MS). Inclusion of as many currently regulated and CCL 3 SVOCs as possible into a single analytical method will reduce monitoring costs by allowing laboratories to perform simultaneous monitoring for compliance under the SDWA and future UCMRs. Because SPE technology has advanced since Method 525.2 was last updated, EPA's National Exposure Research Laboratory (NERL) has investigated updating the method to take advantage of new SPE products, while also expanding the analyte list. The expanded list includes new CCL 3 chemicals, a group of compounds from EPA's National Homeland Security Research Center (NHSRC) and other emerging contaminants. Literature reviews3–7 indicated that many CCL 3 SVOCs as well as analytes already listed in Method 525.2 (over 100) could be analyzed together in a single updated method, while also improving overall method performance. Contaminants evaluated for inclusion in an updated method, Method 525.3, are listed in Table 2.| Alachlor | Atrazine |
| Bis(2-ethylhexyl)phthalate | Endrin |
| Hexachlorobenzene | Heptachlor |
| Hexachlorocyclopentadiene | Heptachlor epoxide |
| Pentachlorophenol | Methoxychlor |
| Chlordane | Simazine |
| Toxaphene | Benzo[a]pyrene |
| Lindane | Bis(2-ethylhexyl)adipate |
| PCBs |
| a Currently in EPA Method 525.2.b Removed from consideration during early investigation due to poor chromatographic response.c Removed from consideration due to incompatibility with preservation technique.d Removed from consideration after the multi-laboratory study, due to sporadic low recoveries. | |
|---|---|
| Chemicals listed on CCL 3 | NHSRC chemicals |
| Acephateb | Chlorfenvinphos |
| Acetochlora | Chlorpyrifosa |
| α-HCHa | Dichlorvos |
| BHAd | DIMP (diisopropyl methylphosphonate) |
| Captand | Methyl parathion |
| Dicrotophosb | Mevinphos |
| Dimethipin | Parathion |
| Disulfotona | Phorate |
| Ethion | Phosphamidon |
| Ethopropa | |
| Fenamiphosd | |
| Metolachlora | Other compounds of emerging interest |
| Molinatea | BHT (butylated hydroxytoluene) |
| Nitrofen | DEET (N,N-diethyl-m-toluamide) |
| o-Toluidinec | |
| Oxyfluorfen | |
| Permethrina | |
| Profenofos | |
| Quinolinec | |
| Tebuconazole | |
| Tribufos | |
| Vinclozolin | |
In addition to expanding the analyte list, one of the goals in updating Method 525.2 was to modify the sample preservation scheme. Tap water samples to be analyzed by Method 525.2 are dechlorinated with sodium sulfite and subsequently acidified with hydrochloric acid. The acidification serves as a microbial inhibitor and to inhibit hydrolysis of some pesticides.2 This scheme is problematic for several reasons. First, the two-step scheme prevents bottles from being sent to the collection site with preservatives inside. Second, the hydrochloric acid, a strong corrosive, presents safety issues during shipping and for sample collection personnel. Finally, the low pH prevents the efficient recovery of triazine-based SVOCs, such as cyanazine.8–10
Another issue to be evaluated was the replacement of C-18 bonded silica sorbents with newer polymeric sorbents. Silica-based sorbents can cause undesirable silanol interactions.11,12 Also, an extremely low pH (∼2) is required to retain pentachlorophenol.13 Finally, C-18 sorbents are not water wettable, and must be carefully conditioned prior to use, and kept wet at all times during sample processing, which could affect data reproducibility. Thus, a number of polymeric sorbents, based on the divinylbenzene (DVB) monomer and the DVB N-vinylpyrrolidone copolymer, were investigated. There has been reported success using DVB-based polymer sorbents to extract and quantitate a wide range of SVOCs, including organochlorinated and phosphorous-containing pesticides, triazines, phenols, as well as the non-polar polychlorinated biphenyls (PCBs) and polycyclic aromatic hydrocarbons (PAHs).14–19
Sensitivity issues with some Method 525.2 components were also addressed by NERL, because several regulated analytes have low EPA regulatory limits. Compounds such as benzo[a]pyrene, heptachlor epoxide, and heptachlor have Maximum Contaminant Levels (MCLs) of 0.2 μg L−1, 0.2 μg L−1, and 0.4 μg L−1, respectively. Also, several CCL 3 compounds, such as disulfoton, profenofos, ethoprop, alpha-hexachlorocyclohexane (α-HCH), and oxyfluorfen, have health reference levels (HRLs) of less than 1 μg L−1.20 While full scan measurements at sub-microgram per liter levels are certainly within modern MS capabilities, it would be easier to reliably meet the sensitivity requirements in selected ion mode (SIM). SIM will also yield lower Lowest Concentration Minimum Reporting Limits (LCMRLs), a statistical estimate of the lowest level at which a single laboratory can meet the specified data quality objectives, for regulated compounds that have an MCL goal (MCLG) of zero. This provides EPA an opportunity to consider lowering the MCL for those compounds. Therefore, the option of performing analyses in SIM for compounds requiring extra sensitivity was investigated.
Finally, NERL has investigated solutions to high quantitative bias for some method analytes that exhibit matrix-induced chromatographic response enhancement (MICRE). This is a phenomenon in which improvements in analyte peak quality are observed in sample extracts when compared to solvent based standards, due to the presence of co-extracted matrix material which binds to thermally active sites in the GC.21 Often, MICRE is a problem associated with food analysis, but it has also been documented in water analysis.22–25 Organophosphorous pesticides are one of the classes of compounds susceptible to MICRE. Several of these pesticides are candidates for inclusion in Method 525.3. The analytes of concern were typically phosphorus-containing compounds, such as mevinphos, dichlorvos, and fenamiphos, as well as pentachlorophenol. In food analysis, additives to sample extracts known as “analyte protectants” are added to samples and calibration standards prior to GC injection. These protectants bind to active sites in the GC injector and column, preventing those sites from affecting sensitive analytes and eliciting a response that is comparable in both standards and matrix-containing extracts. NERL investigated various forms of analyte protection, such as polyethylene glycol (PEG), ethylglycerol, gulonolactone, and D-sorbitol during method development.
2. Experimental
2.1. Chemicals and standards
Laboratory reagent water (LRW) was prepared from tap water using reverse osmosis followed by a Millipore (Billerica, MA) Milli-Q® Ultrapure Gradient A-10 polishing unit. Acetone, methanol (MeOH), dichloromethane (DCM), and ethyl acetate (EtOAc) were purchased in Optima grade from Thermo Fisher Scientific (Waltham, MA). Sodium sulfate (anhydrous), ACS grade was purchased from Thermo Fisher Scientific (Waltham, MA). The sodium sulfate was baked for 4 hours at 400 °C and packed into 12 mL polypropylene column reservoirs with polyethylene frits for use in the removal of water from final extracts. Potassium dihydrogen citrate, a buffering agent used to adjust sample pH to 3.8 to inhibit microbial growth and analyte degradation, was purchased from GFS Chemicals (Powell, OH). Ethylenediaminetetraacetic acid (EDTA), trisodium salt, added to samples to inhibit metal-catalyzed hydrolysis of analytes, and L-ascorbic acid, to reduce free chlorine at the time of sample collection, were purchased from Sigma-Aldrich (St. Louis, MO).Chemical standards, including those carried over from EPA Method 525.2, were obtained in solution and in neat form from Accustandard, Inc. (New Haven, CT), Ultra Scientific (N. Kingstown, RI) and Absolute Standards (Hamden, CT), except for diisopropyl methylphosphonate (DIMP), which was obtained from Cerilliant Corporation (Round Rock, TX), and 13C-penatchlorophenol, which was purchased neat from CDN Isotopes, Inc. (Pointe-Claire, Quebec, Canada). A full list of Method 525.3 analytes, surrogates, and internal standards are provided in Table 3.
| RT (min) | Internal standards, analytes and surrogates | IS ref. # | QI (m/z) | Confirmation ion(s) (m/z) |
|---|---|---|---|---|
| a Confirmation ions may be at or below 30% relative abundance depending upon instrument tune compounds without values do not have qualifier ions of significant relative abundance. | ||||
| 13.21 | Acenaphthene-d10 (IS 1) | 162 | 164 | |
| 16.35 | 13C-Pentachlorophenol (IS 4) | 276 | — | |
| 16.73 | Phenanthrene-d10 (IS 2) | 188 | 160a | |
| 24.29 | Chrysene-d12 (IS 3) | 240 | 236a | |
| 7.24 | DIMP | 1 | 97 | 123 |
| 8.20 | Isophorone | 1 | 82 | 138a |
| 9.13 | 1,3-Dimethyl-2-nitrobenzene (SUR) | 1 | 77 | 134 |
| 9.97 | Dichlorvos | 1 | 109 | 185a |
| 11.21 | HCCPD | 1 | 237 | 235, 239 |
| 11.53 | EPTC | 1 | 128 | 86 |
| 12.44 | Mevinphos | 1 | 127 | 109a, 192a |
| 12.48 | Butylate | 1 | 57 | 146, 156 |
| 12.71 | Vernolate | 1 | 128 | 86 |
| 12.74 | Dimethylphthalate | 1 | 163 | 77a |
| 12.77 | Etridiazole | 1 | 211 | 183 |
| 12.84 | 2,6-Dinitrotoluene | 1 | 165 | 63, 89 |
| 12.87 | Acenaphthylene | 1 | 152 | — |
| 12.88 | Pebulate | 1 | 128 | 57, 72 |
| 13.43 | 2-Chlorobiphenyl | 1 | 188 | 152 |
| 13.45 | BHT | 1 | 205 | 220 |
| 13.47 | Chloroneb | 1 | 193 | 191 |
| 13.69 | Tebuthiuron | 1 | 156 | 171 |
| 13.79 | 2,4-Dinitrotoluene | 1 | 165 | 63, 89 |
| 13.92 | Molinate | 1 | 126 | 55 |
| 14.31 | DEET | 1 | 119 | 91, 190 |
| 14.44 | Diethylphthalate | 1 | 149 | 177a |
| 14.46 | 4-Chlorobiphenyl | 1 | 188 | 152 |
| 14.53 | Fluorene | 1 | 165 | 166 |
| 14.68 | Propachlor | 1 | 120 | 77, 176 |
| 15.00 | Ethoprop | 1 | 97 | 126, 139, 158 |
| 15.03 | Cycloate | 1 | 83 | 55, 154 |
| 15.27 | Chlorpropham | 1 | 213 | 127 |
| 15.34 | Trifluralin | 1 | 264 | 306 |
| 15.66 | Phorate | 1 | 75 | 121 |
| 15.75 | α-HCH | 1 | 181 | 109, 183, 219 |
| 15.81 | 2,4′-Dichlorobiphenyl | 1 | 222 | 152, 224 |
| 15.83 | Hexachlorobenzene | 1 | 284 | 142, 249 |
| 16.08 | Atraton | 2 | 196 | 169, 211 |
| 16.20 | Simazine | 2 | 201 | 173, 186 |
| 16.21 | Prometon | 2 | 225 | 168, 210 |
| 16.28 | Dimethipin | 2 | 54 | 53 |
| 16.29 | Atrazine | 2 | 200 | 215 |
| 16.30 | β-HCH | 2 | 181 | 109, 183, 219 |
| 16.35 | Pentachlorophenol | 4 | 266 | 264, 268 |
| 16.36 | Propazine | 2 | 214 | 58, 229 |
| 16.46 | γ-HCH (lindane) | 2 | 183 | 109, 181, 219 |
| 16.66 | Pronamide | 2 | 173 | 145 |
| 16.69 | 2,2′,5-Trichlorobiphenyl | 2 | 256 | 186 |
| 16.79 | Phenanthrene | 2 | 178 | 152a |
| 16.81 | Chlorothalonil | 2 | 266 | 264, 268 |
| 16.91 | Disulfoton | 2 | 88 | 61a, 97a |
| 16.91 | Anthracene | 2 | 178 | — |
| 17.01 | Terbacil | 2 | 117 | 161 |
| 17.06 | δ-HCH | 2 | 181 | 109, 183, 219 |
| 17.53 | Phosphamidon | 2 | 127 | 72, 264 |
| 17.68 | Acetochlor | 2 | 146 | 59, 162, 223 |
| 17.71 | Metribuzin | 2 | 198 | — |
| 17.73 | 2,4,4′-Trichlorobiphenyl | 2 | 186 | 256 |
| 17.79 | Vinclozolin | 2 | 212 | 124, 285 |
| 17.84 | Methyl parathion | 2 | 109 | 125, 263 |
| 17.87 | Alachlor | 2 | 188 | 160 |
| 17.96 | Simetryn | 2 | 213 | 155, 170 |
| 18.04 | Ametryn | 2 | 227 | 212 |
| 18.05 | Heptachlor | 2 | 100 | 272, 274 |
| 18.10 | Prometryn | 2 | 241 | 184, 226 |
| 18.39 | Terbutryn | 2 | 226 | 170, 185, 241 |
| 18.47 | 2,2′,5,5′-Tetrachlorobiphenyl | 2 | 220 | 290, 292 |
| 18.53 | Dibutyl phthalate | 2 | 149 | — |
| 18.56 | Bromacil | 2 | 205, 207 | 207, 205 |
| 18.72 | Metolachlor | 2 | 162 | 238 |
| 18.76 | Chlorpyrifos | 2 | 97 | 197, 199 |
| 18.84 | Aldrin | 2 | 66 | 79, 263 |
| 18.87 | Cyanazine | 2 | 225 | 68, 172, 198 |
| 18.87 | Dacthal (DCPA) | 2 | 301 | 332 |
| 18.91 | 2,2′,3,5′-Tetrachlorobiphenyl | 2 | 220 | 255, 292 |
| 18.92 | Ethyl parathion | 2 | 109 | 97, 291 |
| 19.01 | Triadimefon | 2 | 208 | 57 |
| 19.27 | Diphenamid | 2 | 72 | 167, 239a |
| 19.30 | MGK 264(a) | 2 | 164 | 66, 111 |
| 19.59 | MGK 264(b) | 2 | 164 | 66, 111 |
| 19.72 | Heptachlor epoxide | 2 | 353 | 81, 355 |
| 19.74 | Chlorfenvinphos | 2 | 267 | 269, 323 |
| 19.85 | 2,3′,4′,5-Tetrachlorobiphenyl | 2 | 220 | 110, 292 |
| 20.27 | trans-Chlordane | 2 | 375 | 237, 272 |
| 20.38 | Tetrachlorvinphos | 2 | 109 | 329, 331 |
| 20.46 | Butachlor | 2 | 176 | 57, 160 |
| 20.54 | Pyrene | 2 | 202 | — |
| 20.58 | cis-Chlordane | 2 | 375 | 373, 377 |
| 20.59 | Endosulfan I | 2 | 241 | 195, 207 |
| 20.65 | trans-Nonachlor | 2 | 409 | 407, 411 |
| 20.76 | Napropamide | 2 | 72 | 100, 128 |
| 21.00 | Profenofos | 2 | 339 | 97, 139, 208 |
| 21.12 | 4,4′-DDE | 2 | 246 | 176, 318 |
| 21.17 | Tribufos (+merphos) | 2 | 57 | 169 |
| 21.23 | Dieldrin | 2 | 79 | 81 |
| 21.25 | Oxyfluorfen | 2 | 252 | 63, 361 |
| 21.26 | 2,3,3′,4′,6-Pentachlorobiphenyl | 2 | 326 | 184, 254 |
| 21.69 | Nitrofen | 2 | 283 | 139, 202 |
| 21.72 | Endrin | 2 | 263 | 81, 281 |
| 21.81 | 2,2′,3,4′,5′,6-Hexachlorobiphenyl | 2 | 360 | 218, 290 |
| 21.90 | Chlorobenzilate | 2 | 251 | 111, 139 |
| 21.91 | 2,3′,4,4′,5-Pentachlorobiphenyl | 2 | 326 | 184, 254 |
| 21.99 | Endosulfan II | 2 | 195 | 207, 241 |
| 22.11 | 4,4′-DDD | 2 | 235 | 165 |
| 22.11 | Ethion | 2 | 231 | 97, 153 |
| 22.40 | 2,2′,4,4′,5,5′-Hexachlorobiphenyl | 2 | 360 | 218, 290 |
| 22.82 | Norflurazon | 2 | 145 | 102, 303 |
| 22.91 | Butylbenzylphthalate | 2 | 149 | 91, 206 |
| 22.91 | Endosulfan sulfate | 2 | 272 | 237, 387 |
| 23.03 | 4,4′-DDT | 2 | 235 | 165 |
| 23.06 | 2,2′,3,4,4′,5′-Hexachlorobiphenyl | 2 | 360 | 218, 290 |
| 23.17 | Hexazinone | 2 | 171 | 83a |
| 23.38 | Di(2-ethylhexyl)adipate | 3 | 129 | 57, 70 |
| 23.39 | Tebuconazole | 3 | 125 | 83, 250 |
| 23.48 | Triphenyl phosphate (SUR) | 3 | 77 | 169, 326 |
| 24.26 | Benzo[a]anthracene | 3 | 228 | 226a |
| 24.37 | Chrysene | 3 | 228 | 226a |
| 24.40 | Methoxychlor | 3 | 227 | — |
| 24.67 | 2,2′,3,4,4′,5,5′-Heptachlorobiphenyl | 3 | 394 | 252, 324 |
| 24.93 | Di(2-ethylhexyl)phthalate | 3 | 149 | 167 |
| 25.79 | Fenarimol | 3 | 107 | 139, 219 |
| 26.64 | cis-Permethrin | 3 | 183 | 163a |
| 26.82 | trans-Permethrin | 3 | 183 | 163a |
| 27.49 | Benzo[b]fluoranthene | 3 | 252 | 126a |
| 27.54 | Benzo[k]fluoranthene | 3 | 252 | 126a |
| 28.22 | Benzo[a]pyrene-d12 (SUR) | 3 | 264 | 132a |
| 28.28 | Benzo[a]pyrene | 3 | 252 | 126a |
| 28.50 | Fluridone | 3 | 328 | 329 |
| 30.72 | Indeno[1,2,3-c,d]pyrene | 3 | 276 | 138a |
| 30.80 | Dibenzo[a,h]anthracene | 3 | 278 | 139a |
| 31.20 | Benzo[g,h,i]perylene | 3 | 276 | 138a |
2.2. Solid-phase extraction procedure and apparatus
(a) SPE cartridge extraction − Experiments utilizing 6 mL SPE cartridges, such as Waters Oasis HLB 500 mg (Catalog # 186000115), Varian Bond Elut C-18 (Cat. # 12102052), Varian Bond Elut-LMS (Cat. # 12255021), Waters RDX (Cat. # WAT047220), and Baker DVB H2O-phobic 200 mg (Cat. # 8109-09), were performed using a Supelco Visiprep™ 24-position vacuum manifold (Sigma-Aldrich, St. Louis, MO). Once inserted onto the manifold, SPE cartridges were conditioned with 5 mL EtOAc, followed by 10 mL MeOH, and rinsed with 10 mL LRW. The sorbent bed was kept wet after the addition of MeOH. Water samples (1000 mL) were fortified with surrogate (SUR) compounds, and then passed through the SPE cartridge under vacuum (−50 kPa) at a flow rate of 5–10 mL min−1 using PTFE Visiprep™ Large Volume Sampler (Sigma-Aldrich, St. Louis, MO) sample transfer lines. Sample bottles were rinsed with 10 mL LRW after sample loading and the rinsate passed through the cartridge. The sample transfer lines were removed and cartridges were dried under vacuum for 10 min. The sample transfer lines were reattached, and 15 mL sample collection vials were placed into the manifold, then placed under partial vacuum (−30 kPa). Sample bottles were rinsed with 5 mL EtOAc, pulled through sample transfer lines, allowed to soak on the cartridge for 1 min, then allowed to pass into the 15 mL collection vial. Next, the bottle rinse, soaking process, and solvent transfer was repeated using 5 mL DCM. The combined extracts were dried by passing through 12 mL or 15 mL polypropylene columns packed with sodium sulfate (10 g), followed by a 5 mL DCM rinse of the original collection vial transferred through the sodium sulfate column, collecting into a second 15 mL collection vial. Extracts were concentrated by nitrogen evaporation (N-EVAP, Organomation Assoc., Berlin, MA) at 40 °C to a volume of slightly less than 1 mL. Extracts were then transferred to 1 mL volumetric tubes, followed by the addition of internal standards (IS), and brought to final volume with EtOAc. Finally, extracts were transferred to GC autosampler vials and analyzed by GC-MS. Fortified LRW samples and fortified field samples were handled as described above, but were fortified with compounds from Table 3 just prior to extraction.(b) SPE disk extraction − Experiments using a 47 mm disk (3M SDB-XC, Catalog # 2240) were performed with a 6-place filter manifold apparatus and a suitable collection tube (40 mL collection vial) inserted to contain the extract. Sample preparation, sorbent conditioning, sample loading, drying time, and elution solvents and volumes were the same as described in the cartridge extraction section, but with an extra final rinse of the filtration reservoir with 5 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOAc:DCM transferred to the collection vial. Sodium sulfate drying and nitrogen evaporation were performed in the same manner described in Section 2.2 (a) SPE cartridge extraction.
:1 EtOAc:DCM transferred to the collection vial. Sodium sulfate drying and nitrogen evaporation were performed in the same manner described in Section 2.2 (a) SPE cartridge extraction.
(c) Baker Speedisk disk extraction − Experiments using JT Baker (Phillipsburg, NJ) Speedisk Hydrophobic DVB extraction disks, Catalog # 8068-06 (regular) and Catalog # 8068-07 (high capacity), were performed with a six-port Expanded Extraction Station (JT Baker Catalog # 8095-06). Sample preparation, sorbent conditioning, sample loading, and elution solvents and volumes were the same as described in the cartridge extraction section, but with only 3 min of vacuum drying after sample loading, and an extra bottle rinse and elution of the Speedisk with 2 mL of acetone transferred to the collection vial (40 mL vials) prior to standard cartridge elution. Sodium sulfate drying and nitrogen evaporation were performed in the same manner described in Section 2.2 (a) SPE cartridge extraction.
2.3. Instrumental analysis
A Thermo (Waltham, MA) Finnigan Trace DSQ Quadrupole GC/MS with an AS3000 autosampler was used in full scan and SIM mode for the GC/MS analysis (electron ionization) of SVOCs during method development. Injections were made in the splitless mode, with a 30 psi pressure pulse. The GC oven was equipped with a 5% diphenyl/95% dimethyl polysiloxane column from Restek (Rxi-5Sil MS, Catalog # 13623). A 1 μL volume was injected into the injection port at 275 °C with a split delay of 1 min. The oven temperature program was as follows: initial temperature of 70 °C, hold for 1.5 min, ramp at 10 °C min−1 to 200 °C, ramp at 7 °C min−1 to a final temperature of 320 °C and hold for 3 min. The GC was operated at a constant carrier gas flow rate of 1.2 mL per min helium. Total run time was approximately 32 min. Data acquisition commenced at about 7 min. Full-scan mass spectrometer detection was set for m/z range of 45–500, with emission current set to 100 μA. Specific retention times, with quantitation and confirmation ions, for each analyte, along with IS and SUR compounds, are available in Table 3. Quantitative analysis of all analytes was performed using internal standard quantitation and a six-point weighted curve, with linear or quadratic fits for all analytes. SIM was performed using multiple retention time windows, with quantitation and qualifier ion dwell-times set to allow for 0.3–0.6 scans per s scan rates. Specific data parameters for SIM analysis have been cited in Method 525.3.263. Results and discussion
3.1. Chromatographic evaluation of CCL 3 compounds
Initial chromatographic evaluation of compounds was performed to ascertain retention times, set up quantitative analysis, and to remove poor performers from further method development. Although tebuconazole, dicrotophos, and acephate were less sensitive than other candidate compounds, they remained in consideration, with experiments performed at higher concentrations (5×) for those compounds.3.2. Evaluation of CCL 3 compound extraction efficiency
Evaluation of CCL 3 compounds on various types of SPE sorbents yielded a wide range of recoveries. NERL's goal was to achieve data quality objectives (DQOs) of 70–130% recovery and replicate relative standard deviation (RSD) values less than 30%. Fig. 1 provides recovery data for selected analytes extracted from replicates (n = 4) of fortified reagent water (4 μg L−1 each analyte, except acephate and tebuconazole which were 20 μg L−1) using various types of SPE media. The data show acceptable efficiency of most compounds under neutral conditions (no preservative) on polymeric sorbents (Waters RDX, Varian Bond Elut-LMS, and Waters HLB). However, acephate and, to a lesser extent, o-toluidine recovery data were poor for all sorbents, under both acidic and neutral conditions. Dimethipin and tebuconazole were not recovered by C-18 sorbent (Varian Bond Elut). Finally, even though quinoline was recovered under neutral conditions, the recovery efficiency suffered greatly under acidic conditions. Due to the poor across-the-board recovery of acephate, coupled with the aforementioned sensitivity issues, it was removed from future experimental studies. Quinoline and o-toluidine were also removed due to poor performance under the final preservation scheme (Section 3.3). | ||
| Fig. 1 Recoveries from fortified reagent water (4 μg L−1 each analyte, 20 μg L−1 tebuconazole) replicates (n = 4) extracted from SPE cartridges with various sorbents (a–d). Control limit bars set at 70% and 130%. | ||
The analytes from EPA Method 525.2 and the remaining CCL 3 compounds, along with a small group of compounds requested for evaluation by EPA's National Homeland Security Research Center (NHSRC), were evaluated together for extraction performance using polymeric-based sorbents, with optimum performance achieved on divinylbenzene sorbents, such as Baker DVB H2O-phobic cartridges, Baker Speedisk DVB disks, Baker High Capacity (HC) Speedisk DVB disks, Waters Oasis HLB cartridges, as well as 3M Empore SDB-XC 47 mm disks. Some compounds were removed for performance reasons. For example, CCL 3 compound dicrotophos exhibited bad chromatographic peak shapes as well as poor recovery from SPE media and thus, was eliminated. Method 525.2 compounds tricyclazole and methyl paraoxon were eliminated due to poor peak shapes and sensitivity. Endrin aldehyde could not be successfully extracted from water samples using the preservation scheme and the SPE media selected for the method and therefore, was eliminated. Fig. 2 shows recovery data from the entire Method 525.3 analyte list (after eliminating problem compounds) in LRW and multiple sources of tap water. Compounds were extracted using Baker HC DVB Speedisk, but similar results were attained for the other aforementioned sorbents. The lone exception was dimethipin, which only met DQOs using Oasis HLB and the Baker HC Speedisk.
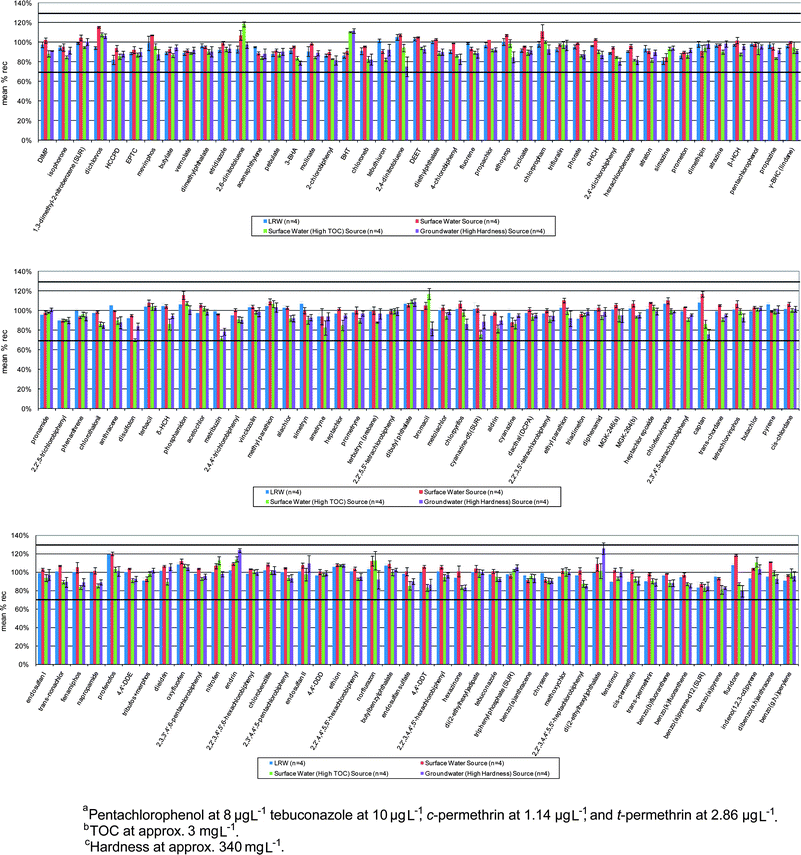 | ||
| Fig. 2 Extraction efficiencies of all Method 525.3 compounds fortified at 2 μg L−1a into 1 L sample replicates (n = 4) of LRW, surface water-source drinking water, surface water-source drinking water with high TOCb, and groundwater-source drinking water with high hardness levelsc. Control limit bars set at 70% and 130%. | ||
3.3. Preservation scheme
To simplify the drinking water collection process, improve triazine compound recovery, and improve safety conditions for collection personnel, a single step preservation scheme mirroring EPA Method 527 (ref. 4 and 27) was evaluated for its feasibility in this current method. Citric acid, EDTA, and L-ascorbic acid, at 9.4 g L−1, 0.35 g L−1, and 0.1 g L−1, respectively, were added to 1 L LRW samples, as well as drinking water samples from groundwater and surface water sources. Samples were stored at 4 °C, protected from light. The majority of compounds met DQOs, with most compound recoveries ranging from 69–121% recovery, with 0–10% RSD.26 Some acid sensitive compounds exhibited initial low recoveries, but data were improved by adding a simple 10 mL LRW rinse to the extraction process prior to vacuum drying the SPE media. The preservatives allowed most analytes to be stored in aqueous samples for at least 14 days under refrigeration. The exceptions were quinoline and o-toluidine, which could not be recovered at acceptable levels using acidic (pH < 4) preservation, and captan, which undergoes hydrolysis under both neutral and acidic conditions.283.4. Analyte protection
Compounds such as dichlorvos, fenamiphos, tebuconazole, and pentachlorophenol have exhibited MICRE in NERL's laboratory studies. MICRE occurs when, in the absence of matrix components, method analytes in calibration solutions are degraded or adsorbed in the GC injector or column, resulting in poor peak shapes and low response.21 Studies involving analyte protectants, compounds added to sample extracts to improve performance of thermally sensitive analytes, were performed with ethylglycerol, gulonolactone, and D-sorbitol at 10 mg, 1 mg, and 1 mg per 1 mL sample, respectively.21,29,30A comparison study of 1 μg mL−1 of analyte mix in EtOAc, and 1 μg mL−1 of analyte mix in an analyte protectant mix (ethylglycerol, gulonolactone, and D-sorbitol) in acetone, was carried out. However, ethylglycerol was immediately excluded from consideration due to mass spectral masking of the front portion of the chromatogram, resulting in the inability to reliably detect several early eluting compounds, such as dichlorvos and hexachlorocyclopentadiene (HCCPD). Also, gulonolactone and D-sorbitol eluted from the GC column as large “blobs” in the middle of the chromatogram, causing mass spectral match failures for some compounds and rendering quantitation difficult (Fig. 3). Concentrations of protectant material were lowered in an attempt to reduce the interferences, but reduced interferences were matched with diminished protective properties. Some compounds exhibited improved peak performance with only a single protectant, but suffered when protectants were combined. For instance, dichlorvos peaks were sharper with gulonolactone added, compared to the addition of all three protectants together. Because many of the analytes of concern benefitted from different protectants, and the aforementioned chromatographic and spectral interferences caused by other compounds, ethylglycerol, gulonolactone, and D-sorbitol were removed from consideration.
 | ||
| Fig. 3 Total ion current (TIC) chromatograms of a blank containing 1 mg mL−1 gulonolactone, a blank containing 1 mg mL−1D-sorbitol, a 1 μg mL−1 standard mix with gulonolactone and D-sorbitol at 1 mg mL−1, and a 1 μg mL−1 standard mix. | ||
The polyether PEG, which has been referenced in various articles as an effective analyte protectant,24,25,31,32 was also used in trial extractions. However, results yielded little improvement in peak shape of susceptible compounds. Also, PEG has been cited to cause rapid deterioration of column performance, which cannot be corrected by simple column trimming.29 Therefore, PEG was also eliminated from consideration.
A commonly used approach to adjust for MICRE in the food chemistry field is the use of matrix-matched calibration standards, which involves the preparation of standards from blank food extracts.33 However, the application to drinking water analysis is a challenge due to different finished drinking water having a wide variation of total organic carbon (TOC) and hardness concentrations. Therefore, the option to prepare standards in solvents from the extraction of LRW was investigated. A set of 1 L LRW samples were preserved and extracted in the standard manner, with all SUR, IS, and standards added immediately before bringing to a final volume of 1 mL. The final product is a set of standards in a matrix extract that mimics actual drinking water extracts, termed “matrix-matched” standards. Initial comparisons of matrix-matched standards and solvent (EtOAc) standards were performed, with an entire calibration curve (0.1–5.0 μg mL−1) of each set analyzed. Results indicated an improved peak shape for several problem compounds, such as dichlorvos, DIMP (an NHSRC compound), s-ethyl dipropylthiocarbamate (EPTC), and fenamiphos (Fig. 4). Peaks from matrix-matched standards exhibited less tailing, peak splitting and fronting than did peaks of solvent-based standards. This phenomenon was particularly true of problem compounds at mid- to low-range concentrations (2 μg mL−1 or less), the range in which peak shape can be critical for maintaining consistent quantitation. For example, dichlorvos, mevinphos, fenamiphos, and oxyfluorfen, when fortified in tapwater at 1 to 2 μg L−1, were recovered at concentrations of 123%, 136%, 137%, and 134%, respectively, when quantitated with calibration standards prepared in EtOAc. However, recoveries of 82%, 84%, 98%, and 109%, respectively, were noted when analytes were quantitated from matrix-matched standards. The latter values coincide with recovery data from analytes normally unaffected from MICRE, and are well within DQOs.
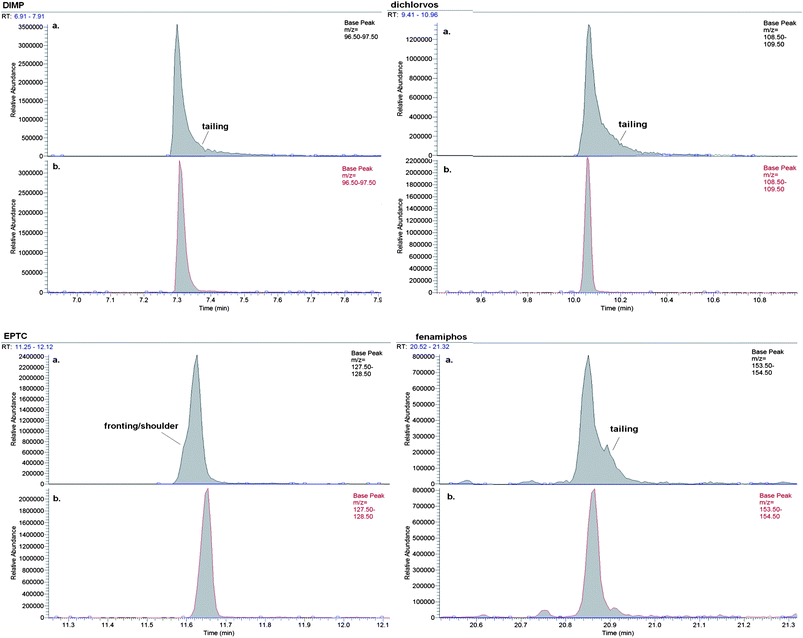 | ||
| Fig. 4 Comparison of extracted quantitation ion currents of select problem analytes at 2 μg mL−1 in (a) EtOAc and (b) matrix-matched solvent. | ||
Trials were performed using matrix-matched standards to quantitate fortified replicates (n = 4) of 1 L LRW samples, drinking water from a surface water source, drinking water from a groundwater source high in mineral content, and drinking water from a surface water source high in total organic carbon (TOC). Fig. 2 contains recovery and precision data of all Method 525.2, CCL 3, and NHSRC compounds considered for inclusion into Method 525.3. Extractions were performed using the Baker H2O-phobic HC DVB Speedisk, but comparable data were obtained for all sorbents listed in the Material and Methods section. The mean recovery of each analyte meets the previously stated DQOs of the research project.
3.5. Selected ion mode option and priority compounds
The EPA MCL of some Method 525.2 compounds, such as benzo[a]pyrene, heptachlor epoxide, and heptachlor, are well below 1 μg L−1, with an MCLG of zero. There are also a number of CCL 3 compounds that have HRL values below 1 μg L−1. Therefore, the option to use SIM, currently not available in Method 525.2, was evaluated for inclusion into Method 525.3.26 Experimental work consisted of a compound list that included many of the regulated Method 525.2 compounds (lower LCMRLs for regulated compounds that have an MCLG of zero provides EPA an opportunity to lower the MCL), and selected CCL 3 and NHSRC compounds from Tables 1 and 2. The group was considered a “priority list” of compounds for SIM evaluation, as well as the analyte group for multi-laboratory comparison studies. Evaluation included analyses of fortified reagent water and various fortified drinking water matrix extractions, performed at concentrations ranging from 0.005 to 0.5 μg L−1 for the priority list compounds.3.6. Multi-laboratory study
Volunteer commercial, instrument vendor, and drinking water utility labs were solicited to evaluate the Method 525.3 priority analytes. The laboratories were instructed to prepare matrix-matched standards, create calibration curves, then analyze and generate accuracy and precision data from replicate fortified reagent water, and laboratory fortified drinking water samples. Also, an LCMRL analysis, described in detail by Winslow et al.,34 was performed in SIM mode. Laboratories were requested to perform this work in the SIM mode, at concentrations (in water) ranging from approximately 10–500 ng L.−1Table 4 contains the precision and accuracy and LCMRL results from the three volunteer labs (randomly labeled Labs ‘A, B, and C’) that completed the project, along with data generated by NERL. Note that reagent water accuracy and precision across all laboratories were from 77.1% to 118% recovery, with RSDs of 0.6% to 30%. Tap water fortified sample accuracy and precision across all laboratories ranged from 77.1% to 128%, with RSDs of 0.5% to 18%. As expected, LCMRL values for compounds with low MCLs, such as benzo[a]pyrene, heptachlor epoxide, and heptachlor were much lower in SIM than full scan. The NERL generated LCMRL concentrations for benzo[a]pyrene, heptachlor epoxide, and heptachlor in full scan were 0.18, 0.27, and 0.11 μg L−1, respectively (data not shown). The values dropped to 0.036, 0.017, and 0.010 μg L−1, respectively, in SIM. The SIM option generated LCMRLs from multiple laboratories lower than all current MCL values for regulated compounds. LCMRL values were also lower than the HRL for all the priority compounds except α-HCH (HRL 0.006 μg L−1). Therefore, the research has successfully demonstrated the option of using SIM for enhanced sensitivity.| Analyte | EPA HRL (H) or MCL (M) (μg L−1) | NERLa | Lab Ab | Lab Bc | Lab Cd | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reagent water (n = 4) | Tap water (n = 4) | LCMRL (μg L−1) | Reagent water (n = 6) | Tap water (n = 7) | LCMRL (μg L−1) | Reagent water (n = 4) | Tap water (n = 4) | LCMRL (μg L−1) | Reagent water (n = 4) | Tap water (n = 4) | LCMRL (μg L−1) | ||||||||||
| % Recovery | % RSD | % Recovery | % RSD | % Recovery | % RSD | % Recovery | % RSD | % Recovery | % RSD | % Recovery | %RSD | % Recovery | % RSD | % Recovery | % RSD | ||||||
| a Samples fortified at 0.1 μg L−1 for most analytes, except PCP (4×) and tebuconazole (5×). Oasis HLB manual SPE, Thermo DSQ GC/MS.b Samples fortified at 0.2 μg L−1 for most analytes, except PCP (4×) and tebuconazole (5×). Baker High Capacity H2O Phobic Speedisk automated SPE, Agilent 5975C GC/MS.c Samples fortified at 0.2 μg L−1 for most analytes, except PCP (4×) and tebuconazole (5×). Oasis HLB automated SPE, Agilent 5975C GC/MS.d Samples fortified at 0.1 μg L−1 for most analytes, except PCP (4×) and tebuconazole (5×). Oasis HLB manual SPE, Agilent 5975C GC/MS.e NC = not calculated. The LCMRL value could not be determined.f Regulated as chlordane.g Cancer HRL.h Non-cancer HRL. | |||||||||||||||||||||
| Alachlor | 2 (M) | 112 | 1.1 | 112 | 3.6 | 0.016 | 93.4 | 10 | 96.6 | 7.4 | 0.006 | 102 | 1.1 | 105 | 2.1 | 0.0014 | 104 | 2.1 | 108 | 0.9 | 0.0056 |
| Atrazine | 3(M) | 118 | 3.6 | 101 | 0.45 | 0.023 | 88.1 | 28 | 110 | 13 | NCe | 101 | 1.1 | 103 | 1.4 | 0.0014 | 104 | 1.4 | 106 | 1.5 | 0.0049 |
| Benzo[a]pyrene | 0.2 (M) | 103 | 2.2 | 104 | 7.6 | 0.036 | 99.4 | 11 | 101 | 6.1 | 0.011 | 81.4 | 2.2 | 81.2 | 1.8 | 0.23 | 94.9 | 2.6 | 95 | 1.8 | 0.003 |
| Chlordane, cis | 2 (M)f | 102 | 2.9 | 97.9 | 3.9 | 0.0073 | 100 | 9.6 | 103 | 6.4 | 0.0091 | 90.0 | 1.7 | 91.5 | 1.7 | 0.05 | 98.3 | 2.7 | 98.6 | 0.9 | 0.00078 |
| Chlordane, trans | 2 (M)f | 99.4 | 5.3 | 102 | 3.7 | 0.0097 | 95.6 | 10 | 99.1 | 6.3 | 0.0078 | 87.6 | 1.7 | 89.4 | 1.8 | 0.094 | 96.7 | 2.6 | 97.2 | 2.1 | 0.00068 |
| Dimethipin | 153 (H) | 108 | 7.4 | 103 | 18 | 0.022 | 102 | 10 | 108 | 5.9 | 0.043 | 96.5 | 0.6 | 96.4 | 2.1 | 0.023 | 93.2 | 18 | 105 | 2.1 | 0.0067 |
| Endrin | 2 (M) | 105 | 3.7 | 107 | 3.3 | 0.014 | 99.0 | 13 | 107 | 8.6 | 0.033 | 105 | 2.5 | 113 | 3.0 | 0.0072 | 104 | 2.2 | 113 | 3.9 | 0.01 |
| Ethoprop | 1.25 (H)g, 0.7 (H)h | 109 | 3.2 | 102 | 3.6 | 0.036 | 103 | 11 | 105 | 5.4 | 0.06 | 105 | 0.9 | 108 | 2.1 | 0.0023 | 102 | 2.9 | 108 | 1.5 | 0.0079 |
| HCCPD | 50 (M) | 108 | 1.2 | 82.8 | 7.3 | 0.014 | 101 | 7.8 | 103 | 3.3 | 0.0061 | 80.5 | 2.4 | 84.2 | 1.1 | 0.21 | 90.4 | 3.7 | 99.6 | 2.0 | 0.00085 |
| HCH, α | 0.006 (H)g, 56 (H)h | 114 | 3.3 | 84.5 | 7.2 | 0.021 | 94.3 | 9.7 | 95.5 | 4.6 | 0.0059 | 100 | 1.0 | 102 | 1.9 | 0.0021 | 99.0 | 3.0 | 98.6 | 2.0 | 0.0036 |
| HCH, γ (lindane) | 0.2 (M) | 110 | 2.0 | 108 | 4.6 | 0.031 | 106 | 8.4 | 110 | 8.5 | 0.014 | 100 | 1.0 | 101 | 1.8 | 0.0022 | 97.9 | 2.3 | 100 | 0.8 | 0.0081 |
| Heptachlor | 0.4 (M) | 99.9 | 3.4 | 85.3 | 5.8 | 0.01 | 94.6 | 9.6 | 96.4 | 4.6 | 0.012 | 95.2 | 2.7 | 99.1 | 3.3 | 0.088 | 95.9 | 2.6 | 102 | 1.4 | 0.0057 |
| Heptachlor epoxide | 0.2 (M) | 103 | 4.6 | 103 | 1.4 | 0.017 | 100 | 9.3 | 104 | 6.1 | 0.0076 | 97.6 | 1.0 | 100 | 1.5 | 0.0014 | 97.7 | 4.2 | 99.2 | 2.2 | 0.0014 |
| Hexachlorobenzene | 1 (M) | 95.3 | 2.1 | 83.8 | 3.3 | 0.014 | 94.3 | 9.6 | 95.4 | 4.0 | 0.005 | 82.0 | 2.0 | 84.2 | 1.1 | 0.16 | 92.5 | 4.0 | 90.5 | 2.3 | 0.0029 |
| Methoxychlor | 40 (M) | 116 | 2.8 | 128 | 1.3 | 0.021 | 104 | 11 | 109 | 5.4 | 0.015 | 110 | 2.8 | 116 | 2.6 | 0.014 | 99.9 | 2.6 | 111 | 0.8 | 0.0035 |
| Oxyfluorfen | 0.478 (H)g, 21 (H)h | 115 | 5.2 | 123 | 4.6 | 0.035 | 106 | 9.9 | 113 | 4.7 | 0.0083 | 108 | 6.3 | 116 | 6.8 | 0.048 | 106 | 2.5 | 116 | 3.4 | 0.0065 |
| Pentachlorophenol | 1 (M) | 96.3 | 6.7 | 94.9 | 0.88 | 0.068 | 102 | 8.8 | 102 | 4.6 | 0.11 | 94.6 | 2.3 | 96.2 | 1.7 | 0.03 | 97.8 | 2.4 | 103 | 1.4 | 0.024 |
| Permethrin, cis | 3.65 (H)g, 1.750 (H)h | 113 | 3.2 | 102 | 5.1 | 0.012 | 96.4 | 9.7 | 100 | 5.8 | 0.011 | 86.6 | 2.3 | 86.4 | 1.8 | 0.043 | 97.8 | 2.8 | 102 | 1.7 | 0.0018 |
| Permethrin, trans | 3.65 (H)g 1.750 (H)h | 104 | 2.0 | 108 | 3.0 | 0.02 | 95.8 | 11 | 99.4 | 5.4 | 0.015 | 83.5 | 2.2 | 83.1 | 1.5 | 0.14 | 96.3 | 2.0 | 102 | 2.0 | 0.0028 |
| Profenofos | 0.35 (H) | 92.0 | 2.3 | 110 | 6.8 | 0.029 | 113 | 11 | 116 | 6.6 | 0.011 | 108 | 1.8 | 112 | 3.1 | 0.016 | 104 | 2.9 | 110 | 2.0 | 0.0049 |
| Simazine | 4 (M) | 101 | 13.0 | 105 | 3.0 | 0.048 | 90.3 | 30 | 121 | 13 | NC | 101 | 1.3 | 102 | 1.1 | 0.0019 | 103 | 1.4 | 105 | 1.7 | 0.0051 |
| Tebuconazole | 210 (H) | 116 | 1.9 | 99.9 | 4.5 | 0.2 | 113 | 11 | 115 | 6.6 | 0.01 | 113 | 1.5 | 118 | 2.4 | 0.041 | 105 | 2.4 | 109 | 1.1 | 0.044 |
| Tribufos | 7 (H) | 101 | 3.5 | 110 | 3.5 | 0.023 | 106 | 9.6 | 107 | 6.5 | 0.027 | 91.0 | 5.2 | 96.4 | 6.7 | 0.12 | 103 | 1.3 | 110 | 1.3 | 0.011 |
| Vinclozolin | 21 (H) | 110 | 2.0 | 104 | 4.2 | 0.0098 | 106 | 8.0 | 106 | 5.5 | 0.0061 | 100 | 1.0 | 102 | 2.0 | 0.0014 | 100 | 2.4 | 102 | 0.8 | 0.0029 |
| PCB congeners (by IUPAC#) | 0.5 total (M) | ||||||||||||||||||||
| 2-Chlorobiphenyl (1) | — | 106 | 1.1 | 95.8 | 4.5 | 0.008 | 98.3 | 8.7 | 99.4 | 4.9 | 0.0013 | 87.1 | 1.8 | 89.6 | 1.1 | 0.097 | 95.2 | 4.1 | 92.8 | 2.4 | 0.0015 |
| 4-Chlorobiphenyl (3) | — | 105 | 1.0 | 96.5 | 3.5 | 0.0073 | 99.0 | 9.1 | 100 | 4.1 | 0.0038 | 86.5 | 1.8 | 88.7 | 1.2 | 0.11 | 94.8 | 3.9 | 92.0 | 2.5 | 0.0021 |
| 2,4′-Dichlorobiphenyl (8) | — | 110 | 7.1 | 90.0 | 4.9 | 0.023 | 89.0 | 10 | 90.3 | 5.0 | 0.0078 | 86.9 | 2.0 | 89.1 | 1.1 | 0.1 | 94.9 | 3.6 | 92.6 | 2.1 | 0.00076 |
| 2,2′,5-Trichlorobiphenyl (18) | — | 106 | 3.0 | 92.7 | 3.2 | 0.013 | 98.4 | 9.2 | 102 | 5.4 | 0.0049 | 87.2 | 1.6 | 88.9 | 1.7 | 0.087 | 94.5 | 3.2 | 93.0 | 2.1 | 0.002 |
| 2,4,4′-Trichlorobiphenyl (28) | — | 105 | 1.7 | 91.4 | 2.1 | 0.014 | 97.8 | 10 | 102 | 5.4 | 0.02 | 82.2 | 1.9 | 82.4 | 1.0 | 0.17 | 94.6 | 3.0 | 92.8 | 1.7 | 0.0027 |
| 2,2′,3,5′-Tetrachlorobiphenyl (44) | — | 108 | 5.7 | 81.6 | 5.2 | 0.025 | 103 | 8.8 | 107 | 6.9 | 0.0031 | 84.8 | 1.4 | 85.0 | 1.6 | 0.13 | 95.6 | 3.4 | 93.8 | 1.8 | 0.00061 |
| 2,2′,5,5′-Tetrachlorobiphenyl (52) | — | 105 | 1.3 | 95.3 | 5.0 | 0.0078 | 96.9 | 9.1 | 101 | 6.5 | 0.022 | 84.7 | 1.5 | 84.9 | 1.5 | 0.14 | 93.0 | 3.2 | 90.9 | 2.5 | 0.0029 |
| 2,3′,4′,5-Tetrachlorobiphenyl (70) | — | 103 | 6.5 | 98.8 | 0.85 | 0.036 | 88.4 | 9.3 | 90.2 | 4.4 | 0.024 | 80.7 | 1.7 | 80.7 | 1.6 | 0.18 | 97.7 | 4.2 | 95.2 | 2.0 | 0.00068 |
| 2,3,3′,4′,6-Pentachlorobiphenyl (110) | — | 99.9 | 4.9 | 97.5 | 3.0 | 0.0092 | 99.8 | 11 | 105 | 6.1 | 0.0088 | 80.3 | 1.5 | 80.1 | 1.0 | 0.21 | 95.7 | 2.4 | 95.6 | 2.0 | 0.00065 |
| 2,3′,4,4′,5-Pentachlorobiphenyl (118) | — | 96.5 | 3.7 | 92.3 | 3.0 | 0.013 | 100 | 9.8 | 104 | 5.4 | 0.00065 | 77.1 | 1.7 | 77.1 | 2.1 | 0.28 | 95.6 | 2.9 | 94.9 | 2.0 | 0.00071 |
| 2,2′,3,4,4′,5′-Hexachlorobiphenyl (138) | — | 96.1 | 4.4 | 96.9 | 3.5 | 0.016 | 102 | 9.6 | 105 | 6.7 | 0.0041 | 79.4 | 1.6 | 79.9 | 2.1 | 0.2 | 93.5 | 3.1 | 93.6 | 2.1 | 0.00058 |
| 2,2′,3, 4′,5′,6-Hexachlorobiphenyl (149) | — | 97.4 | 3.1 | 96.6 | 1.5 | 0.012 | 102 | 11 | 106 | 6.2 | 0.00055 | 79.8 | 1.4 | 80.0 | 2.3 | 0.19 | 94.7 | 3.2 | 94.4 | 2.1 | 0.00056 |
| 2,2′,4,4′,5,5′-Hexachlorobiphenyl (153) | — | 98.7 | 0.79 | 94.5 | 0.86 | 0.013 | 100 | 10 | 103 | 5.5 | 0.0044 | 80.1 | 1.7 | 81.1 | 1.5 | 0.23 | 93.2 | 3.6 | 93.3 | 1.8 | 0.0011 |
| 2,2′,3,4,4′,5,5′-Heptachlorobiphenyl (180) | — | 97.6 | 4.3 | 92.6 | 2.1 | 0.015 | 98.3 | 10 | 100 | 6.5 | 0.0046 | 84.1 | 1.1 | 85.5 | 1.3 | 0.14 | 93.5 | 3.1 | 89.7 | 2.0 | 0.0007 |
4. Conclusion
NERL has updated the existing drinking water Method 525.2 to contain an array of compounds from the most recent CCL and from EPA's NHSRC that meet and exceed the DQOs commonly associated with EPA drinking water methods. The new method update, EPA Method 525.3, contains a preservation scheme that is much more user-friendly to sample collection personnel, and uses SPE sorbents that are more amenable to the extraction of pH-dependent compounds, that will aid in the proper quality control of historically troublesome compounds. Furthermore, the new method update contains a SIM option to provide additional sensitivity, along with an option to use matrix-matched standards to achieve better accuracy for compounds that exhibit MICRE in drinking water matrices.5. Disclaimer
The United States Environmental Protection Agency through its Office of Research and Development funded and managed the research described here. It has been subjected to the Agency's administrative review and approved for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use by the U.S. Environmental Protection Agency.Acknowledgements
Three independent laboratories volunteered to participate in a multi-laboratory study that would demonstrate the method performance in both reagent water and tap water samples. These volunteer laboratories were Underwriters Laboratories, South Bend, IN, Suffolk County Water Authority, Hauppauge, NY, and MWH Laboratories, Monrovia, CA.References
- Drinking Water Contaminant Candidate List 3-Final, Fed. Regist., 2009, 51850–51862 Search PubMed.
- J. W. Eichelberger, J. W. Munch and J. A. Shoemaker, Method 525.2-Determination of Organic Compounds in Drinking Water by Liquid–Solid Extraction and Capillary Column Gas Chromatography Mass Spectrometry, Revision 2.0, 1995, http://www.nemi.gov Search PubMed.
- C. C. Leandro, D. A. Bishop, R. J. Fussell, F. D. Smith and B. J. Keely, J. Agric. Food Chem., 2006, 54, 645–649 CrossRef CAS.
- B. V. Pepich, B. Prakash, M. M. Domino, T. A. Dattilio, D. J. Munch and E. K. Price, Environ. Sci. Technol., 2005, 39, 4996–5004 CrossRef CAS.
- T. L. Potter, L. Marti, S. Belflower and C. C. Truman, J. Agric. Food Chem., 2000, 48, 4103–4108 CrossRef CAS.
- M. B. Riley, J. A. Dumas, E. E. Gbur, J. H. Massey, J. D. Mattice, W. Mersie, T. C. Mueller, T. Potter, S. A. Senseman and E. Watson, J. Agric. Food Chem., 2005, 53, 5079–5083 CrossRef CAS.
- H. Sabik, B. Rondeau, P. Gagnon, R. Jeannot and K. Dohrendorf, Int. J. Environ. Anal. Chem., 2003, 83, 457–468 CrossRef CAS.
- A. A. Boyd-Boland and J. B. Pawliszyn, J. Chromatogr., A, 1995, 704, 163–172 CrossRef CAS.
- R. Ferrari, T. Nilsson, R. Arena, P. Arlati, G. Bartolucci, R. Basla, F. Cioni, G. Del Carlo, P. Dellavedova, E. Fattore, M. Fungi, C. Grote, M. Guidotti, S. Morgillo, L. Müller and M. Volante, J. Chromatogr., A, 1998, 795, 371–376 CrossRef CAS.
- G. A. Smith, B. V. Pepich and D. J. Munch, J. Chromatogr., A, 2008, 1202, 138–144 CrossRef CAS.
- I. Ferrer and E. T. Furlong, Environ. Sci. Technol., 2001, 35, 2583–2588 CrossRef CAS.
- M. C. Hennion, J. Chromatogr., A, 1999, 856, 3–54 CrossRef CAS.
- I. Rodriguez, M. P. Llompart and R. Cela, J. Chromatogr., A, 2000, 885, 291–304 CrossRef CAS.
- M. C. Bruzzoniti, C. Sarzanini and E. Mentasti, J. Chromatogr., A, 2000, 902, 289–309 CrossRef CAS.
- M. Castillo, D. Puig and D. Barceló, J. Chromatogr., A, 1997, 778, 301–311 CrossRef CAS.
- S. B. Hawthorne, S. Trembley, C. L. Moniot, C. B. Grabanski and D. J. Miller, J. Chromatogr., A, 2000, 886, 237–244 CrossRef CAS.
- P. Palmer, L. Wilson, A. Casey and R. Wagner, Environ. Monit. Assess., 2010, 1–13 Search PubMed.
- V. Pichon, M. Charpak and M. C. Hennion, J. Chromatogr., A, 1998, 795, 83–92 CrossRef CAS.
- I. Tolosa, J. W. Readman and L. D. Mee, J. Chromatogr., A, 1996, 725, 93–106 CrossRef CAS.
- Contaminant Information Sheets for the Final CCL 3 Chemicals, Office of Water, 2009, http://www.epa.gov/safewater/ccl/pdfs/ccl3_docs/Final%20CCL%203%20Contaminant%20Information%20Sheets.pdf Search PubMed.
- M. Anastassiades, K. Mastovska and S. J. Lehotay, J. Chromatogr., A, 2003, 1015, 163–184 CrossRef CAS.
- M. Lyytikäinen, J. V. K. Kukkonen and M. J. Lydy, Arch. Environ. Contam. Toxicol., 2003, 44, 0437–0444 CrossRef.
- H. G. J. Mol, M. Althuizen, H.-G. Janssen, C. A. Cramers and U. A. T. Brinkman, J. High Resolut. Chromatogr., 1996, 19, 69–79 CrossRef CAS.
- A. Tanabe, H. Mitobe, K. Kawata and M. Sakai, J. Chromatogr., A, 1996, 754, 159–168 CrossRef CAS.
- A. Tanabe, H. Mitobe, K. Kawata, M. Sakai and A. Yasuhara, J. AOAC Int., 2000, 83, 61–77 CAS.
- J. W. Munch and P. E. Grimmett, EPA Method 525.3: Determination of Semivolatile Organic Chemicals in Drinking Water by Solid Phase Extraction and Capillary Column Gas Chromatography/Mass Spectrometry (GC/MS), 2012, http://www.epa.gov/microbes/ordmeth.htm Search PubMed.
- E. K. Price, B. Prakash, M. M. Domino, B. V. Pepich and D. J. Munch, EPA Method 527: Determination of Selected Pesticides and Flame Retardants in Drinking Water by Solid-Phase Extraction and Capillary Column Gas Chromatography/Mass Spectrometry (GC/MS), US EPA Office of Groundwater and Drinking Water, 2005, http://water.epa.gov/scitech/drinkingwater/labcert/analyticalmethods_ogwdw.cfm Search PubMed.
- N. L. Wolfe, R. G. Zepp, J. C. Doster and R. C. Hollis, J. Agric. Food Chem., 1976, 24, 1041–1045 CrossRef CAS.
- K. Mastovska, S. J. Lehotay and M. Anastassiades, Anal. Chem., 2005, 77, 8129–8137 CrossRef CAS.
- C. F. Poole, J. Chromatogr., A, 2007, 1158, 241–250 CrossRef CAS.
- K. Kawata, T. Asada, K. Oikawa and A. Tanabe, J. AOAC Int., 2005, 88, 1440–1451 CAS.
- K. Kawata, H. Mukai and A. Yasuhara, J. Chromatogr., A, 1995, 710, 243–250 CrossRef CAS.
- D. R. Erney, T. M. Pawlowski and C. F. Poole, J. High Resolut. Chromatogr., 1997, 20, 375–378 CrossRef CAS.
- S. D. Winslow, B. V. Pepich, J. J. Martin, G. R. Hallberg, D. J. Munch, C. P. Frebis, E. J. Hedrick and R. A. Krop, Environ. Sci. Technol., 2006, 40, 281–288 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ay25880c |
| This journal is © The Royal Society of Chemistry 2013 |