A novel DNAzyme-based colorimetric assay for the detection of hOGG1 activity with lambda exonuclease cleavage
Shu-Cheng
Liu
,
Hui-Wang
Wu
,
Jian-hui
Jiang
*,
Guo-Li
Shen
and
Ru-Qin
Yu
*
State Key Laboratory for Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: jianhuijiang@hnu.edu.cn; rqyu@hnu.edu.cn; Fax: +86-731-8822782; Tel: +86-731-8822577
First published on 23rd October 2012
Abstract
The generation of 8-oxo-7,8-dihydroguanine (8-oxoG) in DNA is a common type of DNA damage after exposure to reactive oxygen species or drugs. Human 8-oxoG DNA glycosylase/AP lyase (hOGG1) is a kind of base excision repair enzyme specifically used to repair the base excision of 8-oxoG. In this paper, we develop a novel, simple and sensitive strategy for the detection of hOGG1 activity based on the self-assembly of the active HRP-mimicking DNAzyme coupled with lambda exonuclease (λ exo) cleavage. We designed two DNA oligonucleotides that are fully complementary to each other. One is modified with 8-oxoG, the other contains the G-quadruplex DNAzyme sequence. The two single-stranded DNA (ssDNA) firstly hybridize to form a DNA duplex containing an 8-oxoG. In the presence of hOGG1, the formed DNA duplex is selectively cleaved at the 8-oxoG site, yielding a new DNA duplex with a recessed 5′-phosphate terminus. Upon treatment with λ exo, the 5′-phosphoryl ssDNA of the new DNA duplex is digested by λ exo, releasing the G-quadruplex DNAzyme sequence. After addition of hemin, the G-quadruplex–hemin complex is used as a peroxidase-mimicking DNAzyme, catalyzing H2O2-mediated oxidation of 2,2′-azinobis(3-ethylbenzothiozoline)-6-sulfonic acid (ABTS2−) to generate a colorimetric signal. The activity of hOGG1 is directly related to UV/Vis absorption intensity. The results revealed that the method allowed a sensitive quantitative assay of the hOGG1 concentration with a wide range from 0.05–32 U mL−1 and a low detection limit of 0.01 U mL−1.
Introduction
Many mechanisms lead to the damage of an individual's DNA, either as a result of biological mutagens or as a by-product of normal cellular metabolism.1 DNA damage results in mutation or apoptosis, which may lead to initiation of carcinogenesis.2 The generation of 8-oxo-7,8-dihydroguanine (8-oxoG) in DNA is a common type of DNA lesion, arising from exposure to reactive oxygen species in the cell. The DNA base sequence is permanently altered by the formation of 8-oxoG.3 In addition, the 8-oxoG is easily mutagenic due to mispairing with adenine during replication. However, all human cells have the ability to repair chemically and physically damaged DNA.4 To accomplish this, a category of DNA repair proteins has been evolved. 8-oxoG DNA glycosylase/AP lyase (hOGG1) is a kind of DNA glycosylase produced by human cells and specifically initiates the base excision repair of 8-oxoG.5 To initiate base-excision DNA repair, the DNA glycosylases catalyze excision of damaged bases from the genome.6 DNA N-glycosylase is a special species of DNA glycosylase. The N-glycosylic bond between the target base and deoxyribose is hydrolysed by the DNA N-glycosylases.7 The generation of a free base with an apurinic/apyrimidinic (AP) formulates in DNA.8 The DNA glycosylases have several advantages compared to other DNA repair proteins. Such as, DNA glycosylases are comparatively small monomeric proteins and do not require cofactors for their activity.9 Hence, DNA glycosylases are ideal models for investigating interactions between damaged DNA and proteins. hOGG1 is an 8-oxoguanine DNA glycosylase which has both N-glycosylase and AP-lyase activity.10 The N-glycosylase activity can eliminate damaged purines from duplex strand DNA, yielding an AP site. The AP-lyase can cleave the AP site to produce a 5′ phosphate and a 3′-phospho-α,β-unsaturated aldehyde.11 The expression level of hOGG1 was closely associated with the risk of lung cancer.12 To assay the hOGG1 activity, some traditional methods such as radioactive labeling, gel electrophoresis and HPLC are commonly used.13 All these methods, however, are restrained by the requirement for sophisticated instrumentation, complicated procedures and time-consuming preparation. In order to overcome these limits, it is essential to develop some new methods for the detection of hOGG1 activity.DNAzymes represent catalytically active oligonucleotides that have been applied in catalyzing many chemical and biological reactions. One of the most studied DNAzymes is the hemin–G-quadruplex horseradish peroxidase (HRP) mimicking DNAzyme.14 The hemin–G-quadruplex complex acting as horseradish peroxidase, catalyzing H2O2-mediated oxidation of 2,2′-azinobis(3-ethylbenzothiozoline)-6-sulfonic acid (ABTS2−) to generate a colorimetric signal.15 This DNAzyme has been used for sensing biomolecules by the formation of aptamer–substrate complexes16 or as the output readout signal of logic gate operations.
Herein, we have developed a novel, simple, colorimetric and sensitive strategy for the detection of hOGG1 activity based on HRP-mimicking DNAzyme coupled with lambda exonuclease (λ exo) cleavage. Two fully complementary DNA oligonucleotides are hybridized to form a DNA duplex containing an 8-oxoG. The hOGG1 selectively cleaves the duplex at the 8-oxoG site, yielding a new DNA duplex with a recessed 5′-phosphate terminus. Digestion of the new DNA duplex by λ exo leads to the release of G-quadruplex DNAzyme sequence. The formed G-quadruplex–hemin complex acts as a peroxidase-mimicking DNAzyme, which can catalyze H2O2-mediated oxidation of ABTS2− to generate the colored product ABTS˙− (λmax = 418 nm).17 The activity of hOGG1 is directly related to UV/Vis absorption intensity. The hOGG1 concentration is quantified by monitoring the UV/Vis spectra changes.
Experimental
Reagents and materials
hOGG1, BSA, lambda exonuclease, UDG enzyme, APE1 enzyme and endonuclease IV, 1× NEBuffer2 (50 mM NaCl, 10 mM Tris–HCl, and 10 mM MgCl2, pH 7.9) were obtained from New England Biolabs Ltd (Beijing, China). 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS2−) and 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid sodium salt (HEPES) were purchased from Sigma-Aldrich (St. Louis, USA). H2O2 and hemin were obtained from Shanghai Sangon Biological Engineering Technology & Services Co., Ltd (Shanghai, China). Other chemicals were all of analytical grade and used without further purification. Deionized and sterilized water (resistance > 18.2 MΩ cm) was used throughout the experiments.Oligonucleotide 1 (DNA-1), oligonucleotide 2 (DNA-2) and oligonucleotide 3 (DNA-3) were synthesized and purified by TaKaRa (Dalian, China). The sequences of oligonucleotides used in this work were as follows: DNA-1: 5′-TCT CGA TCC CAA CCC GCC CTA CCC-3′ (the guanine marked in italics was modified with 8-oxoG); DNA-2: 5′-GGG TAG GGC GGG TTG GGA TCG AGA-3′; DNA-3: 5′-TCT CGA TCC CAA CCC GCC CTA CCC-3′.
Instrumentation
UV/Vis absorption spectra were recorded on a Shimadzu UV-2450 spectrophotometer. All optical measurements were taken at room temperature unless indicated.Assay of hOGG1 activity by DNA-based machine
In this assay, 5 μL DNA-1 (10 μM), 5 μL DNA-2 (10 μM), 5 μL NEBuffer2 (10×) and 32.5 μL H2O were firstly incubated at 90 °C for 5 min then slowly cooled to room temperature. BSA (10 μg ml−1 final), 5 units of λ exo and varying concentrations of hOGG1 were added to give a reaction volume of 50 μL and the solution was kept at 37 °C for 40 min. Then, 15 μL of 5 μM hemin, 75 μL of 2× HEPES solution (25 mM HEPES, 200 mM NaCl, 10 mM KCl, 0.05% triton, pH 5.2), and 10 μL H2O were added, and the resulting solution was incubated at room temperature for 30 min. Absorption spectra were recorded at 418 nm within 600 s after the addition of 20 μL of 20 mM ABTS2− and 20 μL of 10 mM H2O2.Enzyme activity assays by gel electrophoresis
To verify the feasibility of this strategy, an electrophoresis experiment was performed. Electrophoresis analysis was carried out in 4% agarose gel by GoldView staining to separate the cleaved products from the substrate. The electrophoresis was carried in 0.5× TBE buffer (45 mM Tris, 45 mM boric acid, 1.25 mM EDTA, pH 7.9) at 100 V for 1.5 h at room temperature. After excitation with a WD-9403F UV device, the resulting gel was imaged with a Canon digital camera.Results and discussion
The principle of the biosensor
Scheme 1 depicts the principle of the biosensor for the assay of hOGG1 activity. In this assay, DNA-1 is modified with 8-oxoG. The complementary DNA-2 is a catalytic DNA oligonucleotide containing a guanine-rich sequence that can exhibit peroxidase activity when complexed with hemin. DNA-1 is fully complementary with DNA-2 and the melting temperature (Tm) of the formed DNA duplex after hybridization to DNA-2 is estimated to be 81.4 °C.18 In the presence of hOGG1, the DNA duplex is selectively cleaved by hOGG1 at the 8-oxoG site, splitting DNA-1 into two parts. The short part of DNA-1, which has low Tm, deliberates from the DNA duplex at 37 °C, yielding a new DNA duplex with a recessed 5′-phosphate terminus. Upon addition of λ exo, the resultant 5′-phosphoryl DNA-1 of the new DNA duplex is digested by λ exo in the 5′ to 3′ direction, releasing the G-quadruplex DNAzyme sequence. After addition of hemin, the G-quadruplex DNAzyme sequence complexes with hemin and the G-quadruplex–hemin complex formed in this way is used as a peroxidase-mimicking DNAzyme, catalyzing H2O2-mediated oxidation of ABTS2− to generate the colored product ABTS˙− (λmax = 418 nm). Whereas, the DNA duplex modified with 8-oxoG cannot be selectively cleaved when no hOGG1 is present. Thus, no G-quadruplex DNAzyme sequence is generated after treatment with λ exo and as a result there is also no existence of G-quadruplex–hemin complex. The ABTS2− cannot be oxidized to generate a colorimetric signal, providing a weak background signal for the assay. Since the activity of hOGG1 is directly related to the UV/Vis absorption intensity, the hOGG1 concentration can be quantified by monitoring the UV/Vis spectral changes. | ||
| Scheme 1 Schematic representation of the principle mechanism of the assay. | ||
Proof of principle experiments were carried out to verify the feasibility of the proposed strategy. Fig. 1A displays UV/Vis absorption spectra of the biosensor obtained in response to hOGG1 and in the control experiment, respectively. As shown in Fig. 1A, in the control experiment, DNA-2 hybridized with the normal DNA-3 to form a DNA duplex without 8-oxoG modification followed by treatment with λ exo and hOGG1. The UV/Vis absorption signal (curve a) slowly increased with time, providing a weak background for the assay. It is indicated that hOGG1 response is not to DNA-3 but the modified 8-oxoG site. The other experiments were performed with modified 8-oxoG DNA duplex. In the presence of λ exo but not hOGG1, we observed a similar signal to that for the control experiment, its UV/Vis absorption signal (curve b) also slowly increased with time. Meanwhile, another similar UV/Vis absorption signal (curve c) was observed in the presence of hOGG1 but without λ exo. However, in the presence of both hOGG1 and λ exo, the UV/Vis signal was obvious different (curve d) and the absorption signal increased sharply with time, which was indicative of the successful formation of G-quadruplex–hemin complex. The experimental results clearly demonstrated the feasibility of the principle.
 | ||
| Fig. 1 (A) UV-Vis absorption spectra of the samples under different conditions: (a) unmodified DNA duplex, λ exo and hOGG1; (b) modified DNA duplex and λ exo; (c) modified DNA duplex and hOGG1; (d) modified DNA duplex, λ exo and hOGG1. The concentrations of λ exo and hOGG1 are 100 U mL−1 and 32 U mL−1, respectively. (B) Electrophoresis analysis image under different conditions, lane I: modified DNA duplex digested by hOGG1 (32 U mL−1); lane II: modified DNA duplex digested by λ exo (100 U mL−1) and hOGG1 (32 U mL−1); lane III: only DNA-2; lane IV: modified DNA duplex digested by λ exo (100 U mL−1). | ||
An electrophoresis experiment was performed to further verify the feasibility of the analytical principle. Fig. 1B displays the typical gel electrophoresis images for different conditions. As shown in Fig. 1B, a bright band was observed in lane III where only DNA-2 was added as a control. It was observed that there was only one bright band with relatively low mobility in lane I where the DNA duplex was treated with λ exo but not hOGG1, indicating that the DNA duplex was kept intact and no DNA-2 was released after treatment with λ exo. This phenomenon was similar to the band in lane IV, when only hOGG1 was added without λ exo. However, after treatment with hOGG1 and λ exo, the DNA duplex was selectively cleaved followed by digestion of λ exo resulting in the release of DNA-2, so we could observe two separated bands in lane II. The electrophoresis results further proved the feasibility of the analytical principle.
Optimization of experimental conditions
The concentration of λ exo is a critical experimental parameter for the assay. Fig. 2 depicts the dependence of A/A0 on the λ exo concentration. As shown in Fig. 2, the A/A0 (A is response signal, A0 is background signal) increased with increasing λ exo concentration with a maximum achieved by using 100 U mL−1 of λ exo. Then, A/A0 declined with increasing λ exo concentration after that. Therefore, the optimal concentration of λ exo was chosen to be 100 U mL−1 and used in subsequent experiments.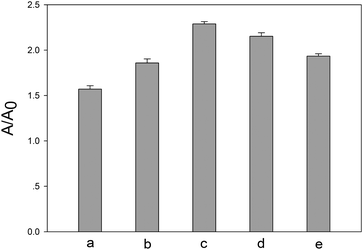 | ||
| Fig. 2 Dependence of the A/A0 values (A is response signal, A0 is background signal) on concentration of λ exo. (a) 50 U mL−1, (b) 80 U mL−1, (c) 100 U mL−1, (d) 120 U mL−1, (e) 150 U mL−1. The concentration of hOGG1 is 32 U mL−1. | ||
The reaction time is another important parameter for the assay. As shown in Fig. 3, A/A0 increased with increasing reaction time till a maximum achieved at 40 min. Thus, we chose 40 min as the optimal reaction time used in subsequent experiments.
 | ||
| Fig. 3 Dependence of the A/A0 values on the reaction time of the two enzymes (32 U mL−1 hOGG1 and 100 U mL−1 λ exo). | ||
Activity of hOGG1 detection
The activity of hOGG1 was quantified by adding varying concentrations of hOGG1 under the optimal experimental conditions. As shown in Fig. 4A, the time-dependent absorbance at 418 nm was observed within 10 min, resulting from the continuous production of ABTS˙− catalyzed by the hemin–G-quadruplex complex. The UV/Vis absorption value gradually increased with the increase of the concentration of hOGG1. This is attributed to the fact that a higher concentration of hOGG1 leads to the yield of more hemin–G-quadruplex complex. It was observed that the hOGG1 assay was quantified from 0.05 to 32 U mL−1. Fig. 4B depicts the absorbance value in the 600 s plotted as a function of the hOGG1 concentration. There was a good linear correlation between the absorbance value and the logarithm of the hOGG1 concentration in the range from 0.05 U mL−1 to 16 U mL−1, as shown in Fig. 4B (inset). The linear relationship is F = 0.4051 + 0.0603log C (F is the absorbance value, log C is the logarithm of the hOGG1 concentration) and the correlation coefficient R2 = 0.9822. The detection limit was estimated to be 0.01 U mL−1 according to the 3σ rule, which was lower than that previously reported.19 The results indicated that the proposed strategy could be used for efficient and highly sensitive detection of hOGG1 activity. | ||
| Fig. 4 (A) Time-dependent absorbance changes upon analyzing different concentrations of hOGG1. The curves a–h are obtained with different concentrations of hOGG1: 0 U mL−1, 0.05 U mL−1, 0.1 U mL−1, 0.5 U mL−1, 2 U mL−1, 8 U mL−1, 16 U mL−1, 32 U mL−1. (B) The absorbance value in the 600 s was plotted as a function of the hOGG1 concentration. Inset: dependence of absorbance value in the 600 s on the logarithm of hOGG1 concentration in the range from 0.05 U mL−1 to 16 U mL−1. Error bars are standard deviations (SD) across three repeat experiments. | ||
Selectivity of the hOGG1 detection
We further investigated the selectivity of the sensing system by using UDG enzyme, APE1 enzyme and endonuclease IV in control experiments. As shown in Fig. 5, all of these three interfering enzymes with the same concentration of 32 U mL−1 failed to generate any obvious UV/Vis absorption changes. However, an obvious UV/Vis absorption change was observed when using 32 U mL−1 of hOGG1. Endonuclease IV is an AP endonuclease which can hydrolyse intact AP sites in DNA, so it will promote the activity of hOGG1.20 Therefore, an obvious UV/Vis absorption change was obtained when endonuclease IV was mixed with hOGG1 in the control experiment. | ||
| Fig. 5 Selectivity of the sensor for hOGG1 compared to other interfering enzymes. The concentrations of hOGG1 and lambda exonuclease are 32 U mL−1 and 100 U mL−1. The other interfering enzymes are 32 U mL−1. The absorbance change (ΔAbs) in 600 s was defined as A − A0. | ||
Conclusion
In summary, we have proposed a simple and sensitive strategy for the colorimetric detection of hOGG1 activity. This novel method is based on the principle that selective cleavage of the DNA duplex containing 8-oxoG by hOGG1 results in a new DNA duplex with a recessed 5′-phosphate terminus and digestion of the phosphorylated DNA duplex by λ exo leads to the release of the G-quadruplex DNAzyme sequence. The method has several advantages: firstly, the preparation of the DNA duplex probe is simple with low-cost; secondly, different concentrations of hOGG1 can be easily distinguished through simple instrumentation; finally, the method exhibits high sensitivity and the detection limit is as low as 0.01 U mL−1. Therefore, the proposed strategy may become a new method of choice for the detection of hOGG1 activity.Acknowledgements
This work was supported by the NSFC (21025521, 21035001, 21190041), National Key Basic Research Program (2011CB911000), CSIRT Program and NSF of Hunan Province (10JJ7002).Notes and references
- S. Loft and H. E. Poulsen, J. Mol. Med., 1996, 74, 297–312 CrossRef CAS.
- M. Michaels and J. Miller, J. Bacteriol., 1992, 174, 6321–6325 CAS.
- R. P. Hickerson, C. L. Chepanoske, S. D. Williams, S. S. David and C. J. Burrows, J. Am. Chem. Soc., 1999, 121, 9901–9902 CrossRef CAS.
- (a) P. Schyman, J. Danielsson, M. Pinak and A. Laaksonen, J. Phys. Chem. A, 2005, 109, 1713–1719 CrossRef CAS; (b) G. Dianov, C. Bischoff, J. Piotrowski and V. A. Bohr, J. Biol. Chem., 1998, 273, 33811–33816 CrossRef CAS.
- C. M. Crenshaw, K. Nam, K. Oo, P. S. Kutchukian, B. R. Bowman, M. Karplus and G. L. Verdine, J. Biol. Chem., 2012, 287, 24916–24928 CrossRef CAS.
- H. E. Krokan, H. Nilsen, F. Skorpen, M. Otterlei and G. Slupphaug, FEBS Lett., 2000, 476, 73–77 CrossRef CAS.
- T. Ellenberger, Chem. Biol., 1995, 2, 351–354 CrossRef CAS.
- H. E. Krokan, R. Standal and G. Slupphaug, Biochem. J., 1997, 325, 1–16 CAS.
- (a) R. K. Singhal, R. Prasad and S. H. Wilson, J. Biol. Chem., 1995, 270, 949–957 CrossRef CAS; (b) G. Dianov and T. Lindahl, Curr. Biol., 1994, 4, 1069–1076 CrossRef CAS.
- S. Boiteux and J. P. Radicella, Biochimie, 1999, 81, 59–67 CrossRef CAS.
- (a) K. W. Caldecott, Nat. Rev. Genet., 2008, 9, 619–631 CAS; (b) L. Wiederhold, J. B. Leppard, P. Kedar, F. Karimi-Busheri, A. Rasouli-Nia, M. Weinfeld, A. E. Tomkinson, T. Izumi, R. Prasad, S. H. Wilson, S. Mitra and T. K. Hazra, Mol. Cell, 2004, 15, 209–220 CrossRef CAS.
- T. Paz-Elizur, M. Krupsky, S. Blumenstein, D. Elinger, E. Schechtman and Z. Livneh, J. Natl. Cancer Inst., 2003, 95, 1312–1319 CrossRef CAS.
- (a) L. H. Lin, S. T. Cao, L. Yu, J. K. Cui, W. J. Hamilton and P. K. Liu, J. Neurochem., 2000, 74, 1098–1105 CrossRef CAS; (b) D. Gackowski, E. Speina, M. Zielinska, J. Kowalewski, R. Rozalski, A. Siomek, T. Paciorek, B. Tudek and R. Olinski, Cancer Res., 2003, 63, 4899–4902 CAS; (c) J. M. Weiss, E. L. Goode, W. C. Ladiges and C. M. Ulrich, Mol. Carcinog., 2005, 42, 127–141 CrossRef CAS.
- P. Travascio, Y. F. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef CAS.
- B. Shlyahovsky, Y. Li, O. Lioubashevski, J. Elbaz and I. Willner, ACS Nano, 2009, 3, 1831–1843 CrossRef CAS.
- (a) D. Li, B. Shlyahovsky, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2007, 129, 5804–5805 CrossRef CAS; (b) K. Sefah, J. A. Phillips, X. L. Xiong, L. Meng, D. V. Simaeys, H. Chen, J. Martin and W. H. Tan, Analyst, 2009, 134, 1765–1775 RSC.
- C. Teller, S. Shimron and I. Willner, Anal. Chem., 2009, 81, 9114–9119 CrossRef CAS.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS.
- (a) E. Cappelli, T. Hazra, J. W. Hill, G. Slupphaug, M. Bogliolo and G. Frosina, Carcinogenesis, 2001, 22, 387–393 CrossRef; (b) B. Liu, X. H. Yang, K. M. Wang and W. H. Tan, Chem. J. Chin. Univ., 2012, 33, 486–491 CAS.
- A. E. Vidal, I. D. Hickson, S. Boiteux and J. P. Radicella, Nucleic Acids Res., 2001, 29, 1285–1292 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |