Simultaneous UV/Vis spectroscopy and surface enhanced Raman scattering of nanoparticle formation and aggregation in levitated droplets
Jonas
Schenk
a,
Lisa
Tröbs
a,
Franziska
Emmerling
a,
Janina
Kneipp
ab,
Ulrich
Panne
ab and
Merwe
Albrecht
*a
aBAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: merwe.albrecht@bam.de
bHumboldt-Universität zu Berlin, Department of Chemistry, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 30th March 2012
Abstract
The formation and growth of hydroxylamine reduced silver nanoparticles were investigated by simultaneous Raman and UV/Vis spectroscopy coupled to an acoustic levitator as a sample holder. Based on the UV/Vis spectra, a two step particle formation mechanism with fast initial formation and adjacent coalescence can be proposed for the reduction of silver nitrate with hydroxylamine. The presence of the analyte adenine during particle formation resulted in differences in the adenine SERS signature compared to experiments, where adenine was added after particle synthesis. It was possible to monitor the adenine and sodium chloride induced aggregation of the nanoparticles and its dynamics based on both the extinction spectra and the SERS data. Correlating the information from the extinction spectra with the SERS intensity, the maximum SERS signals were observed at maximum extinction of the aggregated nanoparticle solution at the Raman excitation wavelength.
Introduction
Acoustic levitation is a technique for contact-free sample handling suitable for all kinds of substances not limited to special sample conditions or shapes. Volumes of levitated droplets are in the μL range, thus larger than volumes in other levitation methods like optical or electrostatic levitation. The levitated volume range depends on the frequency of the ultrasonic wave; lower frequencies enable the levitation of larger volumes. By avoiding container walls, sample contamination can be eliminated and the sample is easily accessible to analytical tools. Acoustic levitation has already been combined to several optical spectroscopy methods such as absorption,1 fluorescence,2,3 and Raman4,5 spectroscopy, or X-ray diffraction.6 In addition, acoustically levitated droplets allow container-less polymerization,7 online reaction monitoring,8 and in situ investigations of concentration induced effects, e.g. aggregation and crystallization.9 The combination of two methods, X-ray diffraction and Raman spectroscopy, has been applied for in situ investigation of crystallization processes in levitated droplets.10 In this study, hyphenation of UV/Vis and Raman spectroscopy for simultaneous measurements is used for the first time to investigate the in situ formation and aggregation behaviour of silver nanoparticles.Silver and gold nanoparticles are widely used as substrates for surface enhanced Raman scattering (SERS). There are different synthesis strategies including chemical reduction,11–13 laser ablation,14,15 or photo reduction.16 For the synthesis of nanoparticles by chemical reduction, several procedures with different reducing agents are known. Among those, the most common ones are sodium citrate,11 sodium borohydride,12 and hydroxylamine hydrochloride.13 Due to the different reduction potential of these reducing agents, the procedures differ in reaction time and temperature, and result in different nanoparticle sizes, shapes, and polydispersity.
For the particle synthesis with the strong reducing agent sodium borohydride a two step mechanism was proposed for both gold and silver nanoparticles.17,18 The fast initial formation of small nuclei is followed by particle growth through coalescence of the nuclei into bigger particles. The formation of gold nanoparticles by reduction with sodium citrate, a weaker reducing agent, is a slower process and a four step mechanism was proposed.19–21 The existing studies indicate a similar formation process for gold and silver nanoparticles for citrate reduction.17,18 To the best of our knowledge, the mechanism of nanoparticle formation by reduction with hydroxylamine has not been described in detail so far.
A large contribution to enhancement in SERS is generated by the so-called electromagnetic enhancement, where both the incident laser light and the Raman scattered light are enhanced due to high local fields around the plasmonic nanostructures. Additional signal enhancement can occur through so-called chemical enhancement e.g., through formation of charge-transfer complexes.22 The SERS effect depends on the nanoparticle size, polydispersity, and presence of nanoaggregates. Therefore, an aggregation agent, for example NaCl, KNO3 or Na2SO4, is often used to improve SERS signal intensity.23 Apart from nanoparticle solutions several other substrates such as immobilized nanoparticles, island films or roughened surfaces can be used for SERS experiments.24
In this study, the formation and growth of hydroxylamine reduced silver nanoparticles as well as influences on their aggregation are investigated by simultaneous Raman and UV/Vis spectroscopy utilizing an acoustic levitator as a sample holder. The lens effect of the spherical surface and the dependence of the optical path length on the droplet volume have to be taken into account in UV/Vis spectroscopy of acoustically levitated droplets. By using a reference droplet and a constant droplet volume a compensation of these effects can be achieved. SERS signal, which is different for isolated and aggregated nanoparticles, serves as an indicator of aggregate formation. Furthermore, the influence of the presence of the analyte, here adenine, during nanoparticle formation is studied.
Experimental
Materials
Silver nitrate (99.8%), hydroxylamine hydrochloride (99.5%), sodium hydroxide (99%) and sodium borohydride (96%) were purchased from AppliChem, sodium chloride (99.99%) from Merck and adenine (99%) from Fluka. All chemicals were used without further purification. For aqueous solutions deionised water (18.2 MΩ) was used. Solutions for the dispenser units were filtered through 0.2 μm filters of regenerated cellulose.Experimental setup
Droplets with typical volumes of 3.5–5 μL were levitated using an acoustic levitator (58 kHz, Tec 5, Oberursel, Germany). The volume range of levitated water droplets ranged from 0.5–8 μL for the used frequency of 58 kHz. The droplets were manually injected into the acoustic levitator with an Eppendorf pipette (0.5–10 μL). For the observation of the droplet volume an IR flash lamp and camera (programmable IR camera, Vision & Control, Jena) were used. An in-house-written program was used to operate the IR camera and to determine the droplet size. The droplet volume was held constant by periodic addition of 0.1 μL water (100 droplets) using a dispenser unit (piezoelectric dispensing system, GeSIM, Großerkmannsdorf). The dispenser unit provides a controllable number of droplets with a volume of ∼1 nL. A scheme of the experimental setup is shown in Fig. 1. | ||
| Fig. 1 Experimental spectroscopy setup for online in situ analysis of reactions in the droplet, including an ultrasonic levitator as the sample holder, Raman and UV/Vis spectrometer, a size determination system and dispensing unit for the investigation and manipulation of the droplet volume. | ||
For the Raman measurements a HeNe laser (633 nm, 35 mW, Melles Griot) was used as an excitation source. The spectra were recorded using an Andor SR-303i spectrometer and a CCD camera (Andor iDus DU420-BV), with a spectral resolution of 5 cm−1. All spectra were recorded with 2 s integration time.
The UV/Vis spectra were recorded using an AvaLight-D(H)-S (Avantes) white light source and an Andor SR-163 spectrograph and a CCD camera (Andor iDus DU420A-BU). The light source and detection unit were coupled to the droplet by fibre optics. The spectra were recorded in the range of 300–620 nm with a spectral resolution of 1 nm. 633 nm short pass filters were used to eliminate the Raman excitation laser from the UV/Vis spectra. For reference measurements a water droplet of 3.5–5 μL was used. Different droplet volumes result in an offset in the UV/Vis spectra. All spectra were recorded with 1 s integration time. For comparison, UV/Vis spectra of larger sample volumes were recorded in quartz cuvettes (Hellma, 10 mm optical path length) using a Jasco V-670 double beam photometer.
The in situ Small Angle X-ray Scattering (SAXS) measurements were conducted at the μSpot beamline at BESSY II synchrotron facility (HZB Berlin). The acoustic levitator was used as a sample holder. The energy was 12.4 kV and the wavelength 1.0 Å. Spectra were taken every 90 s with an exposure time of 60 s and a readout time of 30 s. Colloids prepared in glass vessels were measured with a SAXSess system (Anton Paar). As an excitation source the Cu-Kα emission line was used (1.5406 Å, 40 kV). The scattering curves were fitted using IGOR PRO software with SANS Analysis v6.02 additional toolbox.
Transmission Electron Microscopy (TEM) images were recorded using a JEOL JEM2200FS (JEOL, Eching) transmission electron microscope (200 kV). The sample solutions were deposited on a copper grid and let dry.
The zeta potential of 500 μL samples was measured using a ZETASIZER Nano ZS (Malvern Instruments) with a disposable zeta cell at 25.0 °C with three times 100 measurements each.
Results and discussion
The acoustic levitator serves as a microreactor and allows for online in situ studies of the reaction in small sample volumes. The levitator offers several advantages, in particular the absence of analyte adsorption effects on container walls and optical interference caused by them, which are particularly important for miniaturized systems. On the other hand, evaporation of the solvent and possible influences of the ultrasonic field have to be considered. The experiments carried out for this study can be divided basically into four parts (Fig. 2). In Experiment I the formation of pure nanoparticles was investigated by UV/Vis spectroscopy. The droplet volume was kept constant by periodic addition of water with the disperser unit at early times and allowed to evaporate stepwise afterwards. To gain information on the applicability of the synthesized nanoparticles for SERS measurements, the particles were synthesized either in the presence of adenine (Exp. II) or adenine was added after the synthesis (Exp. III). Additionally, the influence of NaCl as an aggregation agent was investigated (Exp. IV). UV/Vis and Raman spectra were recorded simultaneously in Experiments II–IV and during the evaporation of the solvent. In Fig. 2 the time evolution of the intensity of the plasmon band and SERS intensity is only shown for a constant droplet volume. The SERS intensity is represented by the intensity of the ring breathing mode of adenine at ∼740 cm−1. Examples of SERS spectra of adenine are shown in Fig. 2 top left together with the time evolution, which is marked by a thick black line (1). In the UV/Vis extinction spectrum the positions of the investigated wavelengths are marked: the intensity at the maximum of the plasmon band (2) and the intensity at 620 nm (3). The latter wavelength was chosen to reflect the extinction near the Raman laser wavelength due to interferences of the laser light in the UV/Vis spectra at 633 nm.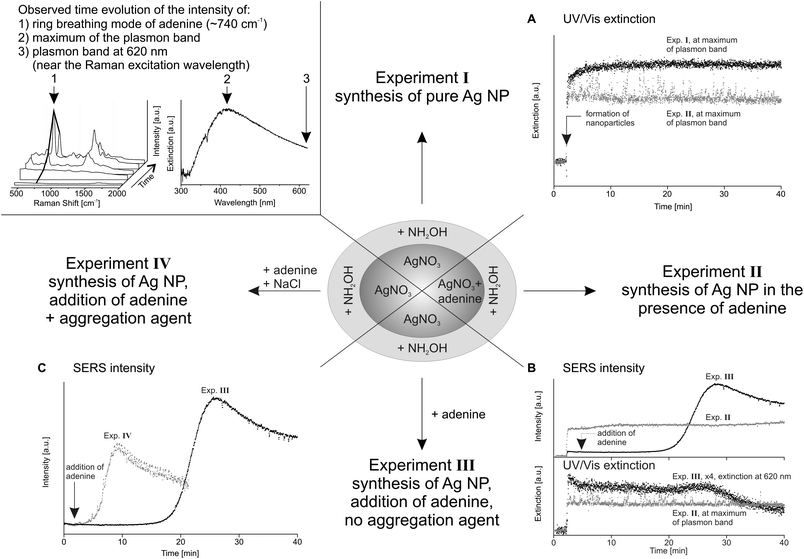 | ||
| Fig. 2 Scheme of the experimental results. Top left: example SERS spectra of adenine and UV/Vis spectrum of silver nanoparticles with marked intensities for the time evolution plots; A: time evolution of the intensity at the maximum of plasmon band of Exp. I (black) and II (grey); B: time evolution of the SERS intensity at ∼740 cm−1 and UV/Vis extinction of Exp. II (grey) and III (black); and C: time evolution of the SERS intensity of Exp. III (black) and IV (grey). | ||
Silver nanoparticle formation and characterization
Silver nanoparticles were synthesized in the levitated droplet by chemical reduction using NH2OH (ref. 4) and NaBH4 (ref. 25) for comparison. Upon adding NH2OH·HCl/NaOH (30 mM/100 mM, 0.4 μL) into a AgNO3 (1.1 mM) containing droplet (3.5 μL) the colour of the droplet changed immediately from colourless to grey and some seconds later to a greyish yellow. For the synthesis of silver nanoparticles by reduction with NaBH4 0.5 μL of AgNO3 solution (2.2 mM) was added to a 5 μL droplet of NaBH4 solution (1.2 mM); the solution immediately turned clear yellow after the short appearance of black schlieren. In situ UV/Vis spectra of the levitated droplet display an immediate (1 s) increase of the silver plasmon band intensity after addition of NH2OH into the AgNO3 solution (Fig. 3, left). The spectra of the NH2OH reduced silver colloids show a broad plasmon band. The maximum of the plasmon band is located at 420 nm 5 seconds after addition of NH2OH and shifted to 411 nm after approximately 25 s. In this time, the FWHM drops from ∼260 nm to ∼160–180 nm. The maximum intensity of the plasmon band, the position of the maximum, and the width of the band were at a constant level after about 30 seconds (see the inset in Fig. 3). Comparison to the reduction with NaBH4 (Fig. 3, right) reveals a very similar time evolution of the plasmon band intensity but the plasmon band has its maximum at 391 nm and is much narrower (FWHM ≈ 50 nm). In the case of NaBH4 reduction, the maximum extinction was also reached after approximately 30 s. The size of the hydroxylamine reduced nanoparticles was determined during synthesis in the levitated droplet using in situ SAXS at the BESSY II synchrotron facility. Evaluation of the SAXS data recorded three minutes after particle synthesis results in nanoparticle diameters of 10–12 nm with a polydispersity of about 33%. The large polydispersity is in agreement with the broad plasmon band (FWHM > 160 nm). | ||
| Fig. 3 UV/Vis spectra recorded during formation of silver nanoparticles in the acoustic levitator. Left: reduction with NH2OH, right: reduction with NaBH4; inset: time evolution of the extinction at the wavelength of the maximum of the plasmon band. Addition of the reducing agent is at 0 s. | ||
For comparison, additional SAXS measurements and TEM measurements were recorded of larger volumes of silver colloids prepared in a glass vessel. These measurements were conducted with one day old colloids and resulted in particle diameters of 50 nm (37% polydispersity, SAXS) and 31 ± 20 nm (TEM). The observed particle sizes of 31–50 nm lie between those published by Leopold and Lendl13 and Papadopoulou and Bell26 with 23–34 nm and 80 nm, respectively. The determined particle sizes of the one day old colloids show a significant larger particle size and still a very high polydispersity compared to the in situ SAXS results.
The size difference can be explained by the difference in time when the particles were investigated. The nanoparticle size was measured by in situ SAXS directly after particle synthesis. In this stage particle growth occurred mainly through coalescence of small nuclei.17 The particle size is smaller compared to the measurements of the one day old colloids, where the particle growth also includes aggregation of particles. The difference in average particle size may therefore be explained by the progression of particle growth and partial aggregation. This can be supported by UV/Vis spectra of a one day old colloid showing a red shift of the plasmon band maximum of about 10 nm compared to spectra recorded directly after the synthesis (data not shown).
Since the plasmon band is detectable within seconds after the addition of NH2OH it is clear that the formation of the particles is a fast process. The colour of the colloid is grey at first and subsequently turning to yellow. This suggests the presence of very polydisperse particles in the beginning of the formation process. Afterwards agglomeration to particles with lower, but still high, polydispersity occurs. These steps are similar to those proposed for the reduction using NaBH4 with fast formation of very small particles in the first step and coalescence in the second step.18 The initial formation of the particles occurs under complete consumption of the ionic silver. Therefore, further particle growth can only be achieved by coalescence of the nuclei. This coalescence step is in contrast to the model of La Mer and Dinegar,27 who proposed particle growth only from the addition of monomers to the particle surface.18,19 In addition, the time evolution of the plasmon band maximum (Fig. 3, inset) shows the same behaviour for the reduction with both reducing agents, an almost linear increase to a constant level of extinction after approximately 30 seconds. Therefore, a similar particle formation mechanism can be proposed for the reduction with hydroxylamine as for the reduction with NaBH4.
The different silver nanoparticle sizes obtained with the two reducing agents indicate that there is still a difference in the formation mechanism. The oxidation of BH4− leads to non-reductive borate species which persist in the particle suspension.18 In contrast, mainly volatile N2 and N2O are produced during the oxidation of NH2OH.28 Since oxidation of NH2OH results in undissolved products which could have a stabilizing effect, this may explain the larger particle sizes and polydispersity of the hydroxylamine reduced particles in comparison to the borohydride reduced particles.
The aggregation behaviour of the NH2OH reduced nanoparticles was first investigated in a droplet with a constant volume. Afterwards, the experiment was carried out with respect to an increasing nanoparticle concentration due to evaporation of the solvent. First, silver nanoparticles were synthesized in a levitated droplet and its volume was kept constant for 40 minutes by periodic addition of 0.1 μL water by a dispenser. Fig. 2A shows the maximum intensity of the plasmon band as a function of time. After the fast nanoparticle synthesis the maximum intensity of the plasmon band slightly increased for about 10 minutes (Fig. 2A, black curve). In the following 30 minutes the extinction spectrum of the colloid remained constant indicating their stability in this time.
After evaporation of a portion of the solvent, droplets with 94%, 63%, and 31%, respectively, of the initial volume were investigated (data not shown). These volumes were held constant for 5 minutes, respectively. The solvent evaporation to 94% of the initial volume did not induce any changes in the extinction spectrum. The FWHM of about 160 nm of the plasmon band at 414 nm remained constant. When the droplet was allowed to evaporate to 63%, the plasmon band broadened to a FWHM of about 200 nm. When the droplet was kept constant at this volume, again no changes in the spectra were observed. The plasmon band showed further broadening (FWHM ≈ 230 nm) upon the volume decrease to 31% of the initial volume, reflecting a wider particle size distribution. Since the initial particle formation is assumed to be complete, this wider size distribution can only be achieved by aggregation. In accord with the assumption that decreasing droplet volume and the resulting increasing nanoparticle concentration have a profound impact on the stability of the nanoparticles, the spectral changes were most pronounced in the droplets of smaller volume. No spectral changes were observed during periods of a constant droplet volume, therefore we conclude that aggregation at a constant volume is absent or only present to a too small extent to be detected in the UV/Vis spectra.
Influence of the analyte on nanoparticle formation and aggregation
For SERS experiments with the synthesized silver nanoparticles, adenine was used as a test molecule (final concentration 1 × 10−5 M). We performed simultaneously both UV/Vis and SERS measurements. In Exp. II adenine was supplied in the initial AgNO3 containing droplet when NH2OH was added to induce particle formation. The above described process of nanoparticle synthesis observed by UV/Vis spectroscopy was also investigated by Raman spectroscopy in the presence of the analyte. As indicator for the SERS enhancement of the synthesized nanoparticles the intensity of the ring breathing mode of adenine (∼730 to 740 cm−1) was plotted over time (Fig. 2B, SERS intensity).Similar to the nanoparticle synthesis in the absence of adenine, the nanoparticle formation in the presence of adenine is a fast process. The plasmon band was detectable within the first seconds after addition of NH2OH (Fig. 2A). Both position and FWHM of the plasmon band are comparable to the experiment in the absence of adenine. In the corresponding in situ Raman spectra SERS signals of adenine were observed in the first spectrum (2 s) after addition of the reducing agent. Therefore, it can be concluded that nanoparticles of a suitable size for SERS enhancement were formed within seconds, which supports the above suggested nanoparticle formation mechanism. The SERS intensity remained generally on a constant level when the droplet volume was kept constant (Fig. 2B). The same was the case for the UV/Vis extinction of the plasmon band at the wavelength of maximum intensity and at 620 nm. Nevertheless, a slight increase in SERS intensity and a slight decrease in the extinction at the maximum of the plasmon band were observed after 40 minutes of constant droplet volume, indicating the beginning of the adenine induced aggregation. The analyte-induced aggregation occurs at much later times than expected for the present nanoparticle and analyte concentrations in experiments, where the analyte is added after particle synthesis.29 In the absence of adenine the nanoparticles were completely stable in this time span. Later, once the droplet was allowed to evaporate, the nanoparticles started to aggregate.
Correlation of SERS intensity and nanoparticle aggregation
In the third experiment, the nanoparticles were generated in the absence of adenine, which was added after nanoparticle synthesis. The time evolution of the plasmon band at the wavelength of 620 nm was chosen to reflect the extinction at the incident Raman excitation wavelength of 633 nm (Fig. 2B, UV/Vis extinction). The time evolution of the SERS intensity of adenine is shown in Fig. 2B (SERS intensity).After nanoparticle generation and addition of adenine it took about 15 minutes until the SERS signal began to increase. Coinciding with the increase in SERS intensity, the extinction at 620 nm was slightly increasing as well, marking the onset of aggregation. The aggregation period took about 10 minutes. Due to the aggregation, the distinct silver plasmon band characteristic of individual nanoparticles disappeared, leading to broad featureless extinction spectra. At one point in the aggregation process (25 min), the extinction at the Raman laser wavelength reached a maximum, coinciding with a strong increase in SERS intensity (Fig. 2B, SERS intensity). Since the extinction at 620 nm had to be employed instead of 633 nm, the curve in Fig. 2B shows a maximum extinction earlier than the SERS intensity. At the time point of maximum SERS intensity the plasmonic properties reached an optimum state, which persisted for only a short time (1–2 min). Once these optimal plasmonic properties were not present anymore the extinction at the laser wavelength decreased, together with the SERS signal intensity.
Because other influences leading to particle aggregation, such as an aggregation agent, are excluded in this experiment, we conclude that we observe adenine induced particle aggregation. The analyte-induced aggregation occurred much faster when adenine was added after particle synthesis, compared to Exp. II. Adenine induced aggregation was already observed for higher concentrations on NaBH4 reduced silver nanoparticles.30 The low stability of Ag nanoparticles reduced by NH2OH was also observed by Canamares et al., who showed aggregation caused by alizarine.31 It should be noted that in general, analyte-induced aggregation is an important factor in SERS, not only in the case of silver nanoparticles.32,33
A strong dependence of the SERS signature on the aggregation of the nanoparticles was observed. Upon evaporation of the solvent a blue-shift of the ring breathing mode of adenine from 735 to 746 cm−1 is visible in the spectrum. Furthermore, the bands at 968, 1029 and 1273 cm−1 decreased in intensity, indicating differences in orientation of adsorbed adenine on the surface upon aggregation of the nanoparticles.
In Experiments II and III, the droplet contained the same substances in the same concentrations, i.e., silver nanoparticles produced by reduction with NH2OH and adenine. The only difference between the experiments was the moment of the addition of adenine. In Exp. III it was added after particle formation, in Exp. II it was present during particle formation. In the corresponding SERS spectra, shown in Fig. 4, pronounced differences between the two experiments were observed. The main differences lie in the spectral region from ∼1250 to 1400 cm−1 and in the relative intensity of the band of the ring breathing mode at ∼740 cm−1.
 | ||
| Fig. 4 SERS spectra of adenine present during particle formation (Exp. II) and added after particle formation (Exp. III, maximum SERS intensity). | ||
SERS spectra of the experiment, where adenine is added after particle formation (Exp III, Fig. 4, black spectrum), correspond to a typical spectrum of adenine (10 ppm, 7.4 × 10−5 M) on hydroxylamine reduced silver colloids at pH 7 measured by Papadopoulou and Bell,26 for which the adenine concentrations lie in the same order of magnitude as in our experiments (10−5 M).
For the experiments, where adenine was present during the process of particle formation (Exp. II), the SERS spectra of adenine (Fig. 4, grey spectrum) show several differences compared to Exp. III. The relative intensity of the ring breathing mode at ∼740 cm−1 is much lower and bands in the region of 1250–1400 cm−1 differ in position and intensity. Comparison of SERS spectra with Raman spectra of polycrystalline adenine and computational studies indicated an upright position of adenine on colloid surfaces via the N7 or the N3 ring atom (see the structure in Fig. 4).34,35 The adsorption of adenine on metal substrates is still controversially discussed in the literature.26,35,36 Intensity differences were reported for the N7–C5 stretching mode at 1333 cm−1 for an Ag electrode and an island film prepared by vacuum deposition of silver, where adenine is supposed to be adsorbed in a tilted orientation, in comparison to Ag colloids, where adenine adsorbed primarily perpendicular to the surface.34 As the most pronounced differences in the SERS spectra of Exp. II and III occur in those bands that are assigned to be especially sensitive to the orientation of the molecule at the surface,34 the differences can be explained with a different orientation of adenine on the nanoparticle surfaces when adenine is present during particle formation.
Furthermore, the pH value and tautomerization,26,35 as well as Ag+ complex formation26,35,37 are effects that can have influences on the adsorption of adenine on the silver substrate.26,34,35 The Ag nanoparticle solution is neutral to alkaline (pH 7–11 measured with indicator paper), also after the addition of adenine, therefore the measured SERS spectra are of neutral or deprotonated adenine. The zeta potential of the nanoparticle solution lies between −50 and −55 mV, comparable to silver nanoparticle solutions produced with other reducing agents in the alkaline pH region.38 In future experiments, additional information on the silver nanoparticles and on the interaction of the particles with molecules from the solution could be gained by X-ray photoelectron spectroscopy (XPS).39
To induce aggregation of the nanoparticles in addition to analyte-induced aggregation, NaCl was used as an aggregation agent in Exp. IV. Controlled aggregation is important, especially for quantitative SERS measurements.29 As the aggregation of nanoparticles upon addition of the analyte strongly depends on the properties and concentration of the analyte, NaCl is often used to aggregate the particles. NaCl was injected together with adenine (final NaCl concentration 10−2 M) into the levitated droplet containing the synthesized nanoparticles. The time evolution of the SERS spectra of Exp. III and IV is shown in Fig. 2C. The aggregation of the nanoparticles is much faster in the presence of NaCl, it starts about 2 minutes after addition of adenine and NaCl. The maximum SERS intensity is reached about 6 minutes later, which is about a third of the time it takes for the SERS intensity in the experiment, without the aggregation agent, to reach its maximum (∼25 min). This demonstrates the possibility to influence the time until the maximum SERS intensity is reached.
Conclusions
The formation and growth of hydroxylamine reduced silver nanoparticles as well as influences on their aggregation were investigated in this study by simultaneous Raman and UV/Vis spectroscopy using an acoustic levitator as a sample holder. Based on the fast formation of the surface plasmon band and the comparison of the reduction of silver ions with sodium borohydride and hydroxylamine, a two step particle formation mechanism with fast initial formation and adjacent coalescence for the reduction of silver nitrate with hydroxylamine is proposed. The detection of SERS spectra immediately after particle formation provides further evidence for the fast formation of nanoparticles. The synthesized nanoparticles are stable within the time in which the droplet volume is held constant. Under evaporation of the solvent, aggregation of the nanoparticles occurs.To investigate the influence of an analyte, the test molecule adenine was either present during particles synthesis or added afterwards. Differences in both aggregation behaviour of the nanoparticles and the adenine SERS signature were observed. While the SERS spectrum of adenine added after particle synthesis is typical for adenine on colloids, the SERS spectra of adenine present during particle formation indicate a different orientation on the particles' surface.
Nanoparticles formed in the presence of adenine show almost no aggregation characteristics. In contrast to this, adenine induces aggregation when added after particle synthesis. The SERS intensity can be correlated with the extent of extinction at the excitation laser wavelength due to the localized surface plasmon of the nanoparticles. Due to accelerated aggregation induced by sodium chloride, maximum extinction and maximum SERS signal are reached earlier.
In summary, we demonstrated that hyphenation of Raman scattering and UV/Vis spectroscopy allows for the investigation of the plasmonic properties of metal nanoparticles during their application of SERS, providing additional information on the interactions between SERS substrate and analyte molecules. Future investigations will use the experimental setup for additional studies of nanoparticle formation in levitated droplets. Concentration-dependent effects like analyte-induced nanoparticle aggregation and solvent evaporation will also be further investigated.
Acknowledgements
The authors are grateful for access to the BESSY facility and would like to thank S. Rolf and A. Sarfraz for their experimental support with the SAXS measurements, I. Gornushkin and J. Riedel for helpful discussions, and I. Dörfel for the TEM measurements.References
- O. Rohling, C. Weitkamp and B. Neidhart, Fresenius J. Anal. Chem., 2000, 368, 125–129 CrossRef CAS.
- S. Santesson, M. Andersson, E. Degerman, T. Johansson, J. Nilsson and S. Nilsson, Anal. Chem., 2000, 72, 3412–3418 CrossRef CAS.
- J. Leiterer, M. Grabolle, K. Rurack, U. Resch-Genger, J. Ziegler, T. Nann and U. Panne, Ann. N. Y. Acad. Sci., 2008, 1130, 78–84 CrossRef CAS.
- N. Leopold, M. Haberkorn, T. Laurell, J. Nilsson, J. R. Baena, J. Frank and B. Lendl, Anal. Chem., 2003, 75, 2166–2171 CrossRef CAS.
- S. Santesson, J. Johansson, L. Taylor, I. Levander, S. Fox, M. Sepaniak and S. Nilsson, Anal. Chem., 2003, 75, 2177–2180 CrossRef CAS.
- J. Leiterer, F. Emmerling, U. Panne, W. Christen and K. Rademann, Langmuir, 2008, 24, 7970–7978 CrossRef CAS.
- J. Leiterer, U. Panne, A. F. Thunemann and S. M. Weidner, Anal. Methods, 2011, 3, 70–73 RSC.
- M. Lopez-Pastor, A. Dominguez-Vidal, M. J. Ayora-Canada, T. Laurell, M. Valcarcel and B. Lendl, Lab Chip, 2007, 7, 126–132 RSC.
- J. Leiterer, W. Leitenberger, F. Emmerling, A. F. Thunemann and U. Panne, J. Appl. Crystallogr., 2006, 39, 771–773 CrossRef CAS.
- M. Klimakow, J. Leiterer, J. Kneipp, E. Rossler, U. Panne, K. Rademann and F. Emmerling, Langmuir, 2010, 26, 11233–11237 CrossRef CAS.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- J. A. Creighton, C. G. Blatchford and M. G. Albrecht, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 790–798 RSC.
- N. Leopold and B. Lendl, J. Phys. Chem. B, 2003, 107, 5723–5727 CrossRef CAS.
- J. Neddersen, G. Chumanov and T. M. Cotton, Appl. Spectrosc., 1993, 47, 1959–1964 CrossRef CAS.
- J. Kneipp, X. T. Li, M. Sherwood, U. Panne, H. Kneipp, M. I. Stockman and K. Kneipp, Anal. Chem., 2008, 80, 4247–4251 CrossRef CAS.
- M. Muniz-Miranda, J. Raman Spectrosc., 2004, 35, 839–842 CrossRef CAS.
- J. Polte, R. Erler, A. F. Thunemann, S. Sokolov, T. T. Ahner, K. Rademann, F. Emmerling and R. Kraehnert, ACS Nano, 2010, 4, 1076–1082 CrossRef CAS.
- D. L. Van Hyning and C. F. Zukoski, Langmuir, 1998, 14, 7034–7046 CrossRef CAS.
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thunemann and R. Kraehnert, J. Am. Chem. Soc., 2010, 132, 1296–1301 CrossRef CAS.
- J. Polte, R. Erler, A. F. Thunemann, F. Emmerling and R. Kraehnert, Chem. Commun., 2010, 46, 9209–9211 RSC.
- J. Polte, M. Herder, R. Erler, S. Rolf, A. Fischer, C. Wurth, A. F. Thunemann, R. Kraehnert and F. Emmerling, Nanoscale, 2010, 2, 2463–2469 RSC.
- A. Campion and P. Kambhampati, Chem. Soc. Rev., 1998, 27, 241–250 RSC.
- N. R. Yaffe and E. W. Blanch, Vib. Spectrosc., 2008, 48, 196–201 CrossRef CAS.
- X. M. Lin, Y. Cui, Y. H. Xu, B. Ren and Z. Q. Tian, Anal. Bioanal. Chem., 2009, 394, 1729–1745 CrossRef CAS.
- B. Vlckova, P. Matejka, J. Simonova, K. Cermakova, P. Pancoska and V. Baumruk, J. Phys. Chem., 1993, 97, 9719–9729 CrossRef CAS.
- E. Papadopoulou and S. E. J. Bell, J. Phys. Chem. C, 2010, 114, 22644–22651 CAS.
- V. K. La Mer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- G. Bengtsson, S. Fronaeus and L. Bengtsson-Kloo, J. Chem. Soc., Dalton Trans., 2002, 2548–2552 RSC.
- M. C. S. Pierre, P. M. Mackie, M. Roca and A. J. Haes, J. Phys. Chem. C, 2011, 115, 18511–18517 CAS.
- S. Basu, S. Jana, S. Pande and T. Pal, J. Colloid Interface Sci., 2008, 321, 288–293 CrossRef CAS.
- M. V. Canamares, J. V. Garcia-Ramos, J. D. Gomez-Varga, C. Domingo and S. Sanchez-Cortes, Langmuir, 2005, 21, 8546–8553 CrossRef CAS.
- V. Joseph, A. Matschulat, J. Polte, S. Rolf, F. Emmerling and J. Kneipp, J. Raman Spectrosc., 2011, 42, 1736–1742 CrossRef CAS.
- K. Kneipp, H. Kneipp, R. Manoharan, E. B. Hanlon, I. Itzkan, R. R. Dasari and M. S. Feld, Appl. Spectrosc., 1998, 52, 1493–1497 CrossRef CAS.
- B. Giese and D. McNaughton, J. Phys. Chem. B, 2002, 106, 101–112 CrossRef CAS.
- M. Muniz-Miranda, C. Gellini, M. Pagliai, M. Innocenti, P. R. Salvi and V. Schettino, J. Phys. Chem. C, 2010, 114, 13730–13735 CAS.
- J. Kundu, O. Neumann, B. G. Janesko, D. Zhang, S. Lal, A. Barhoumi, G. E. Scuseria and N. J. Halas, J. Phys. Chem. C, 2009, 113, 14390–14397 CAS.
- R. Huang, L. B. Zhao, D. Y. Wu and Z. Q. Tian, J. Phys. Chem. C, 2011, 115, 13739–13750 CAS.
- R. A. Alvarez-Puebla, E. Arceo, P. J. G. Goulet, J. J. Garrido and R. F. Aroca, J. Phys. Chem. B, 2005, 109, 3787–3792 CrossRef CAS.
- M. S. Bootharaju and T. Pradeep, J. Phys. Chem. C, 2010, 114, 8328–8336 CAS.
| This journal is © The Royal Society of Chemistry 2012 |