Design and development of a field-deployable single-molecule detector (SMD) for the analysis of molecular markers†
Jason M.
Emory
ac,
Zhiyong
Peng
ac,
Brandon
Young
ac,
Mateusz L.
Hupert
ac,
Arnold
Rousselet
b,
Donald
Patterson
c,
Brad
Ellison
d and
Steven A.
Soper
*abce
aDepartment of Chemistry, Louisiana State University, 232 Choppin Hall, Baton Rouge, LA 70803. E-mail: chope@lsu.edu; Fax: +225 578-3458; Tel: +225 578-1527
bDepartment of Mechanical Engineering, Louisiana State University, 2508 P. Taylor Building, Baton Rouge, LA 70803
cCenter for BioModular Multi-Scale Systems, Louisiana State University, 8000 G.S.R.I. Rd., Bldg. 31000, Baton Rouge, LA 70803
dDepartment of Physics and Astronomy, Louisiana State University, 115 Nicholson Drive, Tower Dr, Baton Rouge, LA 70803
eNano-Bioscience and Chemical Engineering, Ulsan National Institute of Science and Technology, Ulsan, South Korea
First published on 18th October 2011
Abstract
Single-molecule detection (SMD) has demonstrated some attractive benefits for many types of biomolecular analyses including enhanced processing speed by eliminating processing steps, elimination of ensemble averaging and single-molecule sensitivity. However, it's wide spread use has been hampered by the complex instrumentation required for its implementation when using fluorescence as the readout modality. We report herein a simple and compact fluorescence single-molecule instrument that is straightforward to operate and consisted of fiber optics directly coupled to a microfluidic device. The integrated fiber optics served as waveguides to deliver the laser excitation light to the sample and collecting the resulting emission, simplifying the optical requirements associated with traditional SMD instruments by eliminating the need for optical alignment and simplification of the optical train. Additionally, the use of a vertical cavity surface emitting laser and a single photon avalanche diode serving as the excitation source and photon transducer, respectively, as well as a field programmable gate array (FPGA) integrated into the processing electronics assisted in reducing the instrument footprint. This small footprint SMD platform was tested using fluorescent microspheres and single AlexaFluor 660 molecules to determine the optimal operating parameters and system performance. As a demonstration of the utility of this instrument for biomolecular analyses, molecular beacons (MBs) were designed to probe bacterial cells for the gene encoding Gram-positive species. The ability to monitor biomarkers using this simple and portable instrument will have a number of important applications, such as strain-specific detection of pathogenic bacteria or the molecular diagnosis of diseases requiring rapid turn-around-times directly at the point-of-use.
Introduction
While many have demonstrated the utility of fluorescence-based single-molecule detection (SMD) for several different types of molecular assays,1–10 the transitioning of SMD into routine clinical measurements has not occurred to-date in spite of its inherent advantages. The advantages afforded by SMD specifically for molecular assays includes, elimination of processing steps in multi-step assays (for example, eliminates the need for amplification steps, such as PCR), reduces the processing time to realize near real-time readout, exquisite analytical sensitivity due to the use of molecular (i.e., digital) counting, and ultra-low limits-of-detection (single-molecule level). In addition, SMD can generate quantitative data without the need for producing calibration plots when the sampling efficiency of single molecules is near unity.The major bottleneck toward realizing the wide-spread use of SMD into many applications has been limitations imposed by the equipment required to make these measurements, typically consisting of large continuous or pulsed lasers, an extensive array of opto-mechanical components and expensive photon transducers, such as CCDs or channel plates.11,12 In fact, most single-molecule measurements are made using laser confocal microscopes due to the ultra-small probe volume they generate providing high signal-to-noise ratios (SNR) for individual molecules and the excellent light gathering ability of the isotropic emission they can produce when using a high numerical aperture microscope objective.
Another bottleneck for transitioning SMD into routine use is the extensive amount of sample preparation that is required prior to carrying out the fluorescence measurement. For example, clinical samples, such as blood, urine, cerebral spinal fluid, saliva, etc., typically require extensive amount of pre-processing to isolate the target molecules to minimize matrix interferences during the single-molecule measurements. For example, many biomarkers such as DNAs and RNAs, are encased within the cellular and nuclear envelopes and therefore, must be released viacell lysis prior to analysis. Also, once lysed, the targets must be isolated using for example solid-phase extractions to eliminate potential interferences in the downstream processing steps and/or interferences during the fluorescence SMD.
There have been a few reports focused on simplifying some of the operational components associated with SMD, for example miniature power supplies developed by Erickson.13 Giudice et al.also reported a photon counting module using Si avalanche photodiodes14 and several groups have also reported SMD for lab-on-a-chip (LOC) devices with potential field-use applications.15–20 However, the literature is currently devoid of any reports detailing the development of a compact and simple SMD instrument, including the processing electronics, opto-mechanics, excitation source and photon transducer, that can provide processing of input samples and the subsequent detection of single molecules using laser-induced fluorescence for potential point-of-use measurements.
The evolution of equipment such as light emitting diodes, laser diodes, optical fibers, and gradient refractive index lenses has important applications to SMD.21 Laser diodes are available in a wide range of wavelengths and offer reduced sizes compared to other lasers, such as air-cooled argon, Ti-sapphire, or helium-neon lasers. Furthermore, many different optical components allow for the control and shaping of the excitation source to create the necessary photon flux to provide near optical saturation for generating high SNR for SMD. On-chip lenses have also been demonstrated to create high photon densities.22–24 Waveguides are another important optical component that can be integrated into microfluidic devices for both illumination and emission collection for SMD. Recently, Yin and coworkers demonstrated anti-resonant reflecting optical waveguides (ARROW) for single-molecule fluorescence correlation spectroscopy (FCS) in a microfluidic format.16 The compact planar opto-fluidic devices were able to generate small excitation volumes (∼fL) and signals coupled to external optical fibers for both excitation and detection. The ARROWs were fabricated by a sacrificial layer process on a silicon nitride substrate with silicon dioxide layers serving as the core.
The drawback to most of the aforementioned approaches was the increased complexity associated with the fabrication of the fluidic chip making it difficult to realize low-cost disposable platforms appropriate for diagnostic applications. Therefore, simple and low-cost optical components incorporated into the microfluidic chip with minimal fabrication steps would be highly advantageous for clinical diagnostics. Seo et al. demonstrated a 2D planar lens in PDMS to focus light from an LED into a fluidic channel.23 They designed single and multi-element lenses to effectively shape the excitation beam to increase the resulting emission intensity from fluorescent nanospheres. The lenses were fabricated in the same step used to fabricate the microfluidic channels. However, the 2D lenses only focused the light in one dimension. The direct integration of fiber optics into micro-capillary electrophoresis devices has been previously demonstrated in the literature as well.22,25–34
When fibers are integrated into microfluidic devices, they can act as waveguides to deliver excitation light to a defined location of the device in a well-controlled volume, producing high photon irradiances. The illumination volume is set by the core diameter of the fiber optic as well as the acceptance angle of the fiber with typical diameters ranging from 4 μm to 1 mm. Fiber optics can also act as collection optics. In a method called “butt-end fiber coupling,” a fiber was placed against a diffuse light source and collected the light from an area of radius r and solid angle defined by the numerical aperture (NA) of the fiber.35 The maximum collection efficiency of a fiber optic cable is achieved by directly integrating the fiber optic into the microfluidic chip butting the end to the excitation zone. The NAs of optical fibers (NA 0.12 to 0.22) are generally lower than a microscope objective typically used in SMD experiments, but high NA fibers are also available (NAs ∼0.48).
The requirement for a small footprint SMD instrument not only includes the opto-mechanical components, but consideration to the data processing electronics as well. Field programmable gate arrays (FPGA) offer a number of advantages compared to standard printed circuit boards for the processing of electronic signals generated from photon transducers (see Supporting Information, Table S1†).36 The integration of FPGA into microfluidic devices has not been extensively reported, except for one clinical application.37 However, no reports have appeared in which a FPGA was used for SMD.
Another issue associated with the utilization of SMD for clinical applications has been the inability of the SMD equipment to prepare samples prior to the optical measurement. Microfluidics offers the possibility of generating systems appropriate for point-of-use analyses that can provide full automation of the sample processing pipeline.17 Coupling microfluidics to SMD offers the capability to generate sample-processing pipelines that reduce processing time through the elimination of processing steps and eliminate the need for operator intervention. In addition, the small sample volume requirements associated with microfluidics are compatible with SMD. Therefore, the use of SMD coupled to microfluidics can produce systems with unique capabilities that can be used for potential point-of-use applications.
SMD of bacteria based on the use of genomic probes offers the potential for fast, sensitive analysis with minimal amounts of false positive results. For example, Castro et al. demonstrated a technique for the rapid detection of specific nucleic acid sequences in unamplified DNA samples from Bacillus anthracis.38,39 They demonstrated the ability to detect 100 amol of target DNA at a SNR of 3 in 200 s and the assay showed good specificity, even in the presence of an excess of B. globigii. In addition, Peng et al. used single-pair fluorescence resonance energy transfer following a ligase detection reaction to detect bacterial species with high specificity and sensitivity.40 The authors reported the ability to identify Gram(+) from Gram(−) bacteria in 2.6 min with single copy limit-of-detection. While the aforementioned papers are excellent examples of using SMD for the rapid reporting of various bacterial species, the hardware used for the measurements were poised on optical tables and in some cases, used mode-locked lasers and microchannel plates with time-correlated single photon counting electronics for processing the photoelectron events. Thus, the SMD hardware was not conducive to point-of-use operation.
In this paper, we describe the hardware and fluidic components necessary for a molecular analysis system that was configured in a compact and potentially field-deployable format capable of performing SMD for the determination of the Gram positive/negative status of bacteria as an example. The system consisted of the opto-mechanics coupled to a microfluidic chip with the interface consisting of fiber optics integrated to the fluidic chip. The processing electronics were based on a FPGA design. The system could detect fluorescence signatures from single molecules and also, could prepare the sample prior to the SMD.
Experimental
SMD instrument design
The hardware was designed to not only perform the SMD, but also control temperatures for the biochemical reaction. There were five major sections associated with the hardware: (1) photon detection, (2) laser diode control, (3) high voltage, (4) microfluidic channel temperature control, and (5) data acquisition. The components and layout of the system are presented in Fig. 1.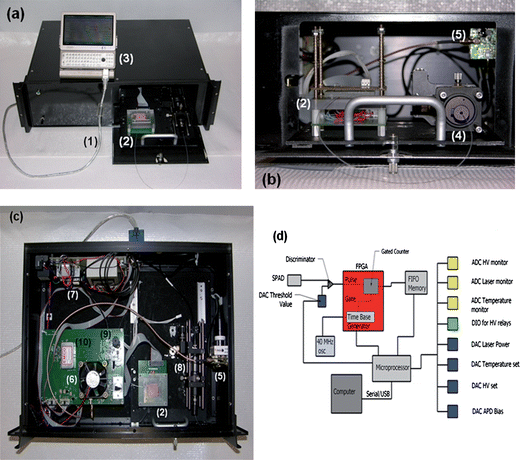 | ||
| Fig. 1 (a) Picture of the compact SMD instrument connected to a mini-computer for data collection and instrument control. (b) Access panel for loading sample into the microfluidic chip and connecting the collection fiber optic to the fiber U-bench, which contained optical fibers and another optical fiber interfaced to the SPAD. (c) Inside the compact SMD instrument showing the arrangement of the VCSEL, SPAD, FPGA and other peripheral electronics. (d) Schematic of the FPGA that was used for data acquisition and single-photon processing. The FPGA counted signals from the SPAD and output information to the first-in first-out (FIFO) memory. (1)–USB interface cable to the controlling computer; (2)–microfluidic chip sitting atop a mounting stage, which is accessed through a drawer that slides out from the main instrument case; (3)–controlling computer; (4)–fiber bench with optical cable connected to the fluidic chip; (5)–SPAD with integrated fiber optic; (6)–cooling fan for the FPGA, which is located underneath this fan; (7)–various power supplies; (8)–fiber bench with optical filters; (9)–VCSEL with integrated fiber; and (10)–electrophoresis power supply to actuate fluids electronkinetically. | ||
An Opnext HL6385DG vertical cavity surface emitting laser (VCSEL) diode was used as the excitation source with a lasing wavelength of 642 nm. To minimize output power fluctuations, a highly stable constant current source was designed to provide up to 280 mA drive current to the laser. The laser diode was connected to an SMA housing for interfacing a fiber optic to it, which also contained a built-in pin diode for laser power monitoring. At 250 mA drive current, the laser output from the coupling fiber optic was ∼24 mW. Conditioning electronics were designed to convert the pin diode output to a 0–5 V signal that was directly proportional to the laser diode output power. An optical fiber was coupled to the SMA housing using an SMA connector and was a 10/125 μm (SMF-28-10, Thorlabs) single-mode fiber. The distal end of this fiber was sealed using epoxy glue into a guide channel embossed into the fluidic chip.
The photon counting system used a PCDMini single photon avalanche diode, SPAD (SensL, Cork, Ireland), that was directly coupled to a fiber optic (200/230 μm BFH48-200, ThorLabs). The PCDMini measured 1½′′ × 1½′′ and had an integrated Peltier cooler. The PCDMini possessed a high single photon quantum efficiency between 650–700 nm (>23%) and an extremely low dark count rate (∼10 cps). The maximum acceptable count rate of the SPAD was determined to be 5M cps (See Supporting Information, Figure S1†).
Collection of the fluorescence photons was achieved using a high NA (0.48) multimode fiber (200/230 μm BFH48-200, Thorlabs) that was sealed within the fluidic chip and oriented at 90° with respect to the excitation fiber. The distal end of the collection optical fiber was spliced into an OFR fiber port (PAF-X-5, Thorlabs) for free beam-to-fiber coupling that allowed placement of the appropriate optical filters for the emission and then coupling back into the fiber optic cable interfaced to the SPAD. In this way, the fiber connected to the SPAD did not need to be replaced every time the fluidic chip was changed.
To minimize dead time and conserve board space, the photoelectrons generated from the SPAD were processed using a custom programmed field programmable gate array (FPGA). The FPGA (XCR3256XL-12TQ44I, Xilinx Inc., San Jose, CA) was a surface mount 144-pin device programmed using the ISE Webpack software, version 7.1. The design used a JTAG interface for in-circuit programming so that the FPGA code could be loaded or modified on the target board without the need to remove the FPGA chip for updating. The SPAD produced a TTL pulse that was sent to the FPGA, which was used for processing the TTLs from the SPAD. A diagram of the electronic components including the FPGA operation is shown in Fig. 1(d). The FPGA was integrated into the SMD instrument as a single-photon counter with a unique dual gated counter configured in a “ping-pong” format to give virtually zero dead time (50 ns) between output counts to the first-in first-out (FIFO) memory.
Finally, communication between the microcontroller and the analog system used 12-bit A/D and D/A converters via an I2C serial bus. Data transmission between the control board and the computer was via the standard USB port. Custom software was written in-house using LabView and installed on a mini computer (OQO, Marlton, NJ) for end user control.
Microfluidic chip fabrication
Fig. 2(a) shows the design of the microfluidic chip, which consisted of a single channel that had a width of 50 μm and a depth of 120 μm. The excitation fiber conduit, which was embossed into the fluidic chip, was 120 μm wide with a depth of 120 μm. The collection conduit was 220 μm wide and 220 μm deep. The excitation and detection geometry is shown in Fig. 2(b), which consisted of a four-way intersection.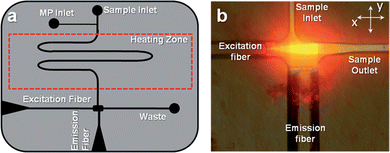 | ||
| Fig. 2 (a) Design of the polymer-based microfluidic chip with integrated fiber optics for delivering excitation light to the chip and collecting the resulting emission. The fibers were placed in guide channels embossed into the chip to allow exact placement during chip assembly and were oriented at 90° with respect to each other. The chip also contained a backside heater to control the temperature for hybridization-based assays. (b) Fluorescence image of the field-of-view of the excitation and emission fibers showing the intersection of the optical paths, which defined the probe volume that was determined to be 98 pL. The chip was filled with Alexa Fluor 660 dye to generate the necessary image. | ||
A detailed description of the microfluidic chip fabrication protocol is described elsewhere41 and a brief description is given in the Supporting Information.† The microfluidic channels were enclosed by thermal fusion bonding a thin (0.25 mm) PMMA coverslip (GoodFellow, Oakdale, PA) to the embossed PMMA substrate following insertion and gluing of the fiber optics to the substrate. For thermal fusion bonding, the microfluidic chip assembly containing the fibers was clamped between two glass plates and placed in a convection oven at 107 °C for 20 min.
Chemicals and materials
Borate buffer was prepared by dissolving the desired amount of sodium borate (Sigma Chemical) into nanopure water secured from a Barnstead Nanopure Infinity System (Model D8991, Dubuque IA). The pH (pH 8.5) was adjusted by the addition of concentrated HCl. The buffer was diluted to a final concentration of 50 mM and filtered with a 0.2 μm filter before use. Bovine serum albumin (BSA) from Sigma was added to the buffer (0.1 mg mL−1) to reduce surface non-specific interactions. AlexaFluor 660 and Dark Red fluorescent FluoSpheres (diameter = 0.2 μm) were both purchased from Invitrogen (Eugene, Oregon). The fluorescent spheres were sonicated and diluted in the borate buffer to yield the desired concentration. The fluorescent dye was diluted in buffer to yield a final concentration of 2.0 fM. Concentrations were selected to keep the occupancy probability below 1.0% to minimize the probability of photon bursts arising from ≥2 molecules occupying the probe volume simultaneously.
Oligonucleotides used to probe for the bacterial genomes and generate molecular beacons (MBs) were purchased from Integrated DNA Technologies (Coralville, Iowa) with a custom sequence (5′-![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) AAAGCCTGACGGAGCAACGCCGCGTGAGTGATGA
AAAGCCTGACGGAGCAACGCCGCGTGAGTGATGA![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) -3′, where the underlined section designates the complementary stem sections of the MB). The 5′ end was modified with a TYE 665 fluorescent dye and the 3′ end was modified with Iowa Black RQ-Sp quencher. The MB sequence was designed to probe for the presence of DNA encoding for ribosomal RNA (rRNA) related to the Gram(+) gene.42 The MBs were diluted in 50 mM borate buffer with BSA to a concentration of 0.5 nM. Two bacterial strains, Staphylococcus aureus subsp. Aureus (ATCC 700699) and Escherichia coli (ATCC 700926) were investigated in this study and the genomic DNA for each strain was acquired from ATCC (Manassas, VA).
-3′, where the underlined section designates the complementary stem sections of the MB). The 5′ end was modified with a TYE 665 fluorescent dye and the 3′ end was modified with Iowa Black RQ-Sp quencher. The MB sequence was designed to probe for the presence of DNA encoding for ribosomal RNA (rRNA) related to the Gram(+) gene.42 The MBs were diluted in 50 mM borate buffer with BSA to a concentration of 0.5 nM. Two bacterial strains, Staphylococcus aureus subsp. Aureus (ATCC 700699) and Escherichia coli (ATCC 700926) were investigated in this study and the genomic DNA for each strain was acquired from ATCC (Manassas, VA).
Flow velocity modeling
A computational fluid dynamic (CFD) simulation was run using ANSYS Fluent 12.0 software. The general-purpose preprocessor for CFD analysis was done with Gambit 2.0, which created quad element meshing with 80,000 nodes. The input volume flow rate for these simulations was 0.05 mL/h.Results and discussion
The diameter of the excitation optical fiber coupled to the microfluidic chip as well as the fiber's divergence served in determining the irradiance and the probe volume size. Reductions in the core diameter of the excitation fiber led to an increase in the irradiance producing better SNR for the single-molecule measurements, but also reduced the excitation volume, consequently, a reduction in the single-molecule sampling efficiency.43 On the other hand, increasing the diameter of the collection fiber along with its numerical aperture and reductions in its distance from the emitting source (i.e., single molecule), increased the amount of fluorescence processed by the system during the residence time of the molecule within the excitation volume.The effective field-of-view of the collection fiber overlaid with the excitation fiber's field-of-view should define the probe volume. Ideally, this volume should be minimized to reduce noise generated from scattering photons produced from the solvent; however, larger collection fibers allowed for more efficient gathering of the fluorescent photons as noted above. To visualize the field-of-views of the excitation fiber and collection fiber, we filled the microfluidic device with Alexa Fluor 660 and launched laser light into both fibers. We then collected two images using a microscope equipped with a CCD camera; (1) Fluorescence generated by the excitation fiber only; and (2) fluorescence generated by the collection fiber only. An overlay image was also produced from images (1) and (2) (see Fig. 2(b)). The effective probe volume was defined by the overlap area of the resulting fluorescence produced from both fiber optics. The cross section of the excitation fiber image produced a diameter of 10 μm due to its low divergence because this was a single mode fiber (cross sectional area = 7.8 × 10−7 cm2). With this cross section, the 24 mW output power from the optical fiber generated an irradiance of ∼1023photon cm−2 s−1. This is a reasonable fluence because it generated an excitation rate, ka (ka = σI, σ is the absorption cross section, cm2, and I is the laser fluence, photons cm−2 s−1) near optical saturation (1/τf, where τf is the fluorescence lifetime of the dye).44
The probe volume was calculated from the 1/e2 diameter of the laser emanating from the fiber (∼25 μm) and the collection fiber (observation length = 200 μm) and was determined to be 9.8 × 10−11 L or 98 pL. This probe volume was used to select the concentration for the SMD experiments so that the occupancy (molecules/probe volume) probability could be adjusted to minimize contributions of double occupancy within the data stream.
The probe volume was configured at a right angle with respect to the sample input into the probe volume with the two fiber optics meeting at a four-way junction to keep the emission fiber close to the excitation fiber to improve the fluorescence collection efficiency. Due to the irregular shape of this detection geometry, we performed simulations to map the flow vectors as the fluid moved through the probe volume and subsequently into the waste reservoir (see Fig. 2(b)). The simulation results indicated a decrease in the linear velocity as the solution entered the wider channel region as expected. The velocity through the probe volume showed minor linear velocity changes; however, the paths through the probe volume were distinct. We categorized these different paths into three types; (1) perpendicular to the excitation axis; (2) diagonal to the excitation axis; and (3) parallel to the excitation axis. We overlaid the CFD simulation results with the trapezoid-shaped (black outline, see Fig. 3(a)) probe volume. The corresponding path lengths across the probe volume for these 3 types were 50, 75, and 200 μm, respectively. These differences may result in different transit times for molecules adopting a trajectory as it moves through the probe volume. The consequence of this is that a broad range of photon burst peak heights would be generated even when operating under a condition where photobleaching is negligible.45
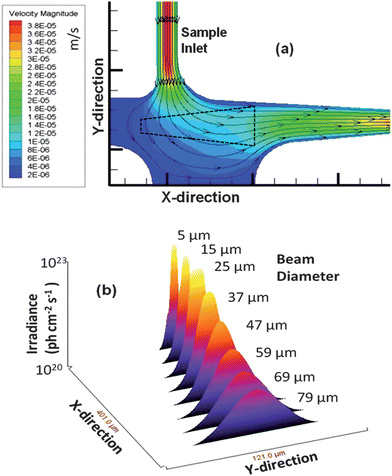 | ||
| Fig. 3 (a) Simulation of the flow velocities and flow vectors of the fluid as it moved from the input channel into the detection zone (units are m s−1). The simulation was run using Fluent software with quad element meshing and 80,000 nodes performed in Gambit. An outline of the probe volume as determined from Fig. 2(b) is shown as well (black dotted line). (b) 3D surface plot of the detection zone versus the irradiance experienced by single fluorescent entities as they traverse through the probe volume. The irradiance decreased as the beam expanded and thus, single fluorescent entities traveling along the edges of the Gaussian intensity profile show reduced photon fluxes. | ||
The distribution of the irradiance as a function of channel position was also plotted versus the irradiance (photon cm−2 s−1) and is shown in Fig. 3(b) with 8 cross sections taken. As can be seen, moving away from the terminal end of the excitation fiber, the irradiance decreased, which will reduce the SNR in the single-molecule measurement. From these results, it was apparent that the perpendicular flow path resulted in the best SNR of the three flow paths due to the higher irradiance it afforded. Additionally, the perpendicular path occurred closer to the emission fiber terminal end and was centered over this fiber, which would result in a higher collection efficiency of the resulting fluorescence photons.
To evaluate various operational parameters associated with this SMD instrument, we used fluorescent microspheres as the probes due to their bright fluorescence signatures resulting from a high load of dye per bead and the lack of photobleaching they demonstrate. A dilute solution of microspheres (3.1 × 106 particles/mL) was used, which had a single particle occupancy probability of 0.3 based on the measured probe volume. The spheres were pumped through the microfluidic channel using a syringe pump at a flow rate of 0.05 mL h−1 (linear flow rate ∼0.096 cm s−1 at 120 μm × 120 μm channel cross section). Fig. 4(a) presents a trace of the photon bursts versus time for these microspheres as well as a blank. We were interested in understanding the distribution in the amplitudes of these bursts and whether the probe volume shape, which was defined by the overlap of the excitation and collection fibers, as well as the various paths taken by the molecules (or microspheres) would affect the variance in this distribution, especially when compared to conventional confocal excitation geometries.
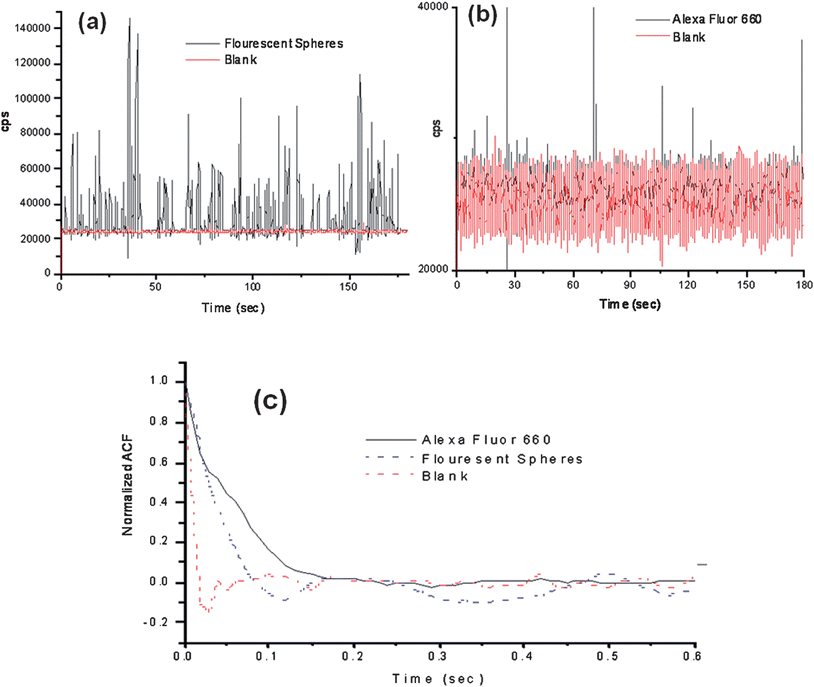 | ||
| Fig. 4 Photon burst data collected using the compact, field-deployable SMD instrument. The red trace shows the blank and the black trace is the data with fluorescent beads or dye seeded into the buffer. (a) Plot of photon bursts generated from fluorescent microspheres. (b) Single AlexaFluor 660 dye molecule burst data. (c) Autocorrelation analysis was performed on the blank, fluorescent spheres and AlexaFluor 660 dye molecule solutions. The fluorescent microspheres and the AlexaFluor dye molecules showed transit times of 49 ms and 53 ms, respectively, using the same flow rate. | ||
For typical confocal laser-induced fluorescence (LIF) single-molecule detectors, the collimated laser beam is tightly focused to a diffraction limited spot with the 1/e2 beam waist defining the excitation volume and whose intensity distribution has a Gaussian profile. When a single fluorescent molecule is brought into this excitation volume, a photon burst is produced due to repetitive cycling of the fluorophore from the ground to excited state with subsequent relaxation back to the ground state accompanied by photon emission. The magnitude of the burst is directly proportional to the local photon density experienced by the molecule during its passage through the laser. The distribution pattern of the photon burst amplitudes can thus serve as an indicator of the intensity profile the molecules take through the excitation volume. In order to examine if the irregular shape of the probe volume arising from the dual optical fiber configuration had any effect on the photon burst distribution of single-molecule events, we ran an experiment by hydrodynamically driving fluorescent beads through the microchannel and histogramming the burst amplitudes to produce a photon burst distribution using a confocal LIF SMD instrument described elsewhere46 and compared these results to the dual-fiber arrangement.
Photon burst distribution plots for both the confocal and dual-fiber SMD instruments were built from the raw trace data. In the single particle detection using the confocal setup, the fluorescent beads were diluted to a concentration of ∼6 × 105 particles/mL to keep the occupany probability low and the solution was driven through the microchannel at a flow rate to give a similar linear velocity as that for the dual-fiber arrangement (∼0.096 cm s−1). The trace data for the photon bursts from the single fluorescent beads detected using the confocal setup is shown in Fig. 5(a). In order to discriminate real photon burst events from background fluorescence fluctuations (i.e., false positives), a threshold level of 3,000 cps was used. A histogram was constructed from this photon burst data and is shown in Fig. 5(c). In this plot, the photon burst magnitude was binned with an increment of 5,000 cps above the threshold level and each bin was normalized to the total number of photon burst events. We found that the photon burst distribution was best fit to a single exponential function (see Fig. 5(c)). The reduced χ2 value of the least square fit was 9.5 × 10−4 and the R2 value was 0.97. From the actual trace data, the average photon burst intensity for the confocal excitation volume was 11,000 ± 12,400 cps. However, we should note that the distribution depicted in Fig. 5(c) represents a truncated Gaussian due to the fact that events were only counted when their maximum exceeded the threshold condition.
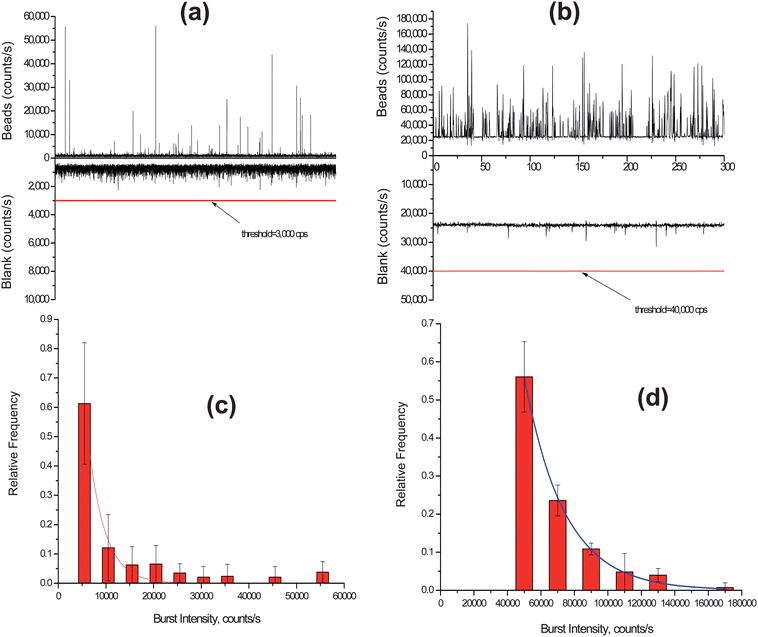 | ||
| Fig. 5 (a) Trace data of photon bursts detected using a confocal LIF setup for the fluorescent spheres. A threshold level of 3,000 cps was set to discriminate true single particle events from background fluorescence fluctuations. (b) Trace data of photon bursts detected using the compact SMD instrument for the fluorescent spheres. In this case, a threshold level of 40,000 cps was set to discriminate single particle events from background fluorescence fluctuations. (c) Histogram of photon burst intensities constructed from the data set in (a) using the confocal LIF setup. The accepted photon bursts were compiled into bins of 5,000 cps. (d) Histogram of photon burst intensities from data in (c) using the compact SMD instrument. The data in this case were accumulated into bins of 20,000 cps. | ||
The photon burst trace data of the fluorescent beads detected using the dual-fiber instrument is shown in Fig. 5(b). A threshold condition of 40,000 cps was set as the discriminator threshold in this case to minimize false positive events. A histogram was built from the photon burst trace data and is shown in Fig. 5(d). The photon burst intensity distribution was also best fit to a single exponential function with a reduced χ2 of 1.12 × 10−4 and a R2 value of 0.997. The average photon burst intensity was 65,800 ± 23,700 cps.
Using the histogram data depicted in Fig. 5(c) and (d), we overlaid a Gaussian function on these exponential functions and from these distributions, determined the standard deviations and relative standard deviations (RSD) for the Gaussian distributions. For the confocal system, the standard deviation was 2,970 cps with an RSD of 27% while for the dual fiber arrangement, the standard deviation was 23,880 cps with an RSD of 37%.
The average photon burst intensity for the dual-fiber system was higher than that of the confocal setup due to the fact that the residence times of the fluorescent microspheres in the dual fiber arrangement were longer than the confocal system (transit time for dual fiber arrangement ≈ 49 ms; transit time for confocal arrangement ≈ 15 ms). However, the relative standard deviation of the burst distribution was larger for the dual-fiber arrangement because of the multiple paths the beads can travel when moving through the excitation volume (see Fig. 3(a)).
Single-fluorophore detection
The SMD system was also analyzed for its ability to detect single-fluorophore molecules (see Fig. 4(b)). In this case, we chose Alexa Fluor 660 due to its high quantum yield and favorable photostablity.47 The dye was diluted to a concentration of 2.0 fM, which corresponded to a single-molecule occupancy probability of 0.12. The solution was pumped through the microfluidic chip at the same volume flow rate as that of the microspheres (0.05 mL h−1). The fluorescent dye showed a drop in SNR compared to the microspheres, which was anticipated due to the high load (1.1 × 105fluorescein equivalents per microsphere) of fluorescent dyes on each microsphere. A discriminator level was set by using a blank sample with no fluorescent molecules. The discriminator level was set such that no signal from the blank exceeded the discrimination level keeping the false positive rate near 0. The single-molecule data showed photon burst signals above the background when a discrimination level of 32,000 cps was selected for the Alexa Fluor dye. From the data trace shown in Fig. 4(d), 48 single-molecule photon burst events were detected.The autocorrelation functions were derived for the blank, fluorescent spheres and the Alexa Fluor dye solutions and are shown in Fig. 4(c). The differences in the autocorrelation function (ACF) from the blank and the fluorescent samples indicated that single molecule events were being detected due to the presence of a non-random component in the ACF. Analysis of the full width at half height of the non-random component in the ACF yielded transit times of 49 ms for the microspheres and 53 ms for the fluorescent dyes. The ACF did not show evidence of the longer transit time components (78–208 ms) associated with some of the flow paths shown in Fig. 3(a); however, the ACF is a weighted average with the weighting factors emanating from the number of events as well as their intensity (i.e., number of fluorescent photons comprising the burst).44 In addition, the transit time is not just a function of the flow velocity of the particles and the size of the probe volume, but also the bleaching lifetime,12,45 and therefore, these longer transit time components may not have been observed due to bleaching artifacts and/or molecules following paths that produce lower numbers of fluorescent photons due to the lower irradiance (see Fig. 3(b)). However, the transit time calculated using the ACF did show close agreement with that calculated from the linear flow velocity and the average 1/e2 radius of the probe volume.
Gram(+) bacterial detection using single MBs
A calibration curve was constructed from oligonucleotides ranging in concentration from 0.5 to 10 fM that were prepared by adding different concentrations of target DNA sequences (synthetic complementary DNA sequence to the loop section of the MB) to a 0.5 nM solution of the MB. The MB and DNA targets were incubated for 5 min and then loaded into a 500 μL syringe and pumped through the optical fiber chip at 0.01 mL h−1 (see Fig. 6). Peaks above the discriminator threshold were counted as events and divided by the time of the experimental run (1 min) with the number of events plotted versus the concentration to construct a calibration curve, which is shown in Fig. 7(a). The data points were fit to a linear function (y = 3 × 10−16x + 4.0 × 10−16) with an R2 value of 0.97. Table 2 in the Supporting Information provides information on the calculation of the sampling efficiency (13.6%) for this system as well as the detection efficiency.†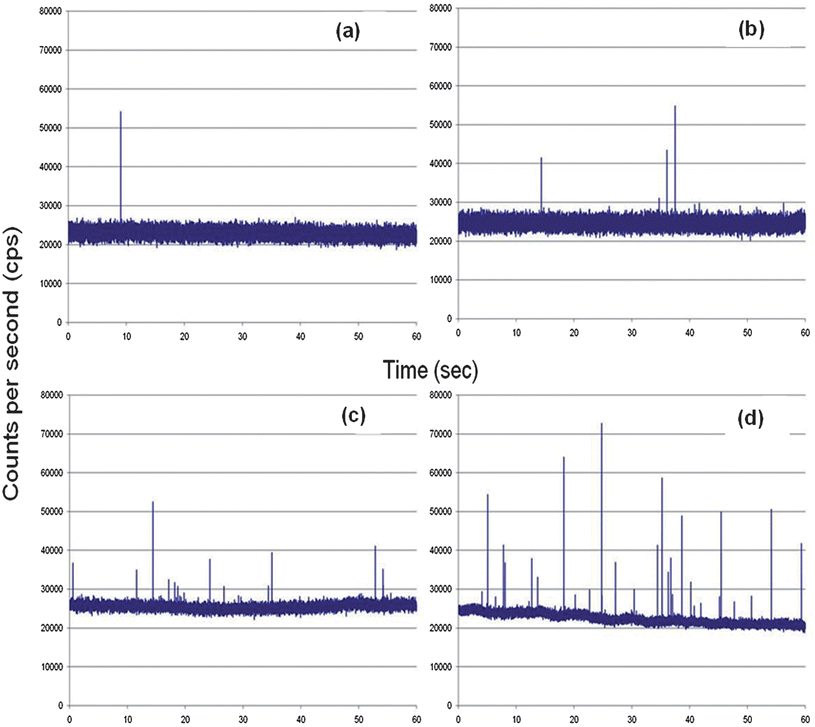 | ||
| Fig. 6 Complementary DNA was mixed with 0.5 nM of the MBs and pumped through the microfluidic chip at 0.01 mL/h. The target concentrations used in this case were; (a) 5.0 × 10−16 M; (b) 1.0 × 10−15 M; (c) 5.0 × 10−15 M; and (d) 1.0 × 10−14 M. | ||
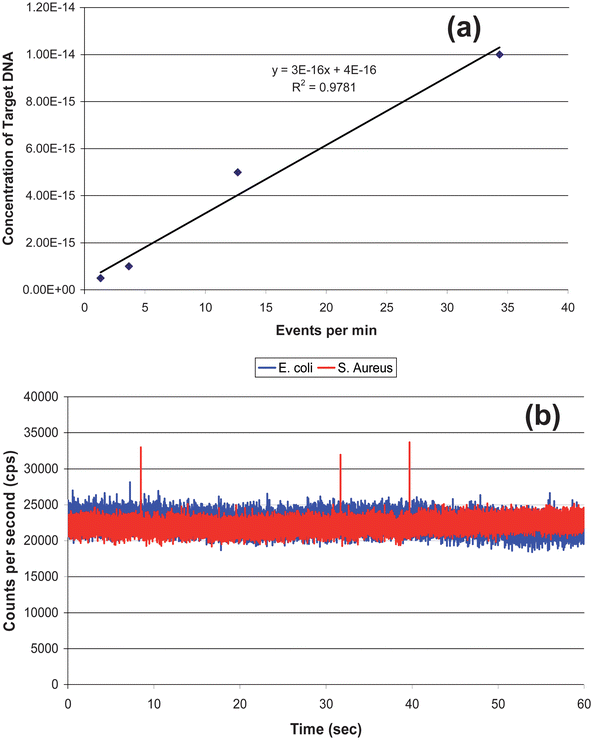 | ||
| Fig. 7 (a) Calibration curve generated from the data shown in Fig. 6. The data points were fit to a linear function, y = 3 × 1015 x − 0.9291, with R2 = 0.97. (b) rDNAextracted from 2,000 S. aureuscells (Gram (+)) and mixed with the 0.5 nM MB solution. As a control, DNA from E. coli (Gram(−)) was extracted and mixed with the MB solution as well. The S. aureus showed 3 events above the discrimination level whereas the E. coli showed no events above this level. | ||
The identification of S. aureus was then performed using DNA extracted from 2,000 cells, which was then mixed on-chip with a 0.5 nM solution of the MBs in 50 mM borate buffer (pH 8.5). The solutions were pumped through the chip at 10 μL h−1 and maintained at a temperature slightly below the Tm of the loop/DNA complement to minimize formation of mismatches. As a negative control, DNA from E. coli was also extracted and mixed with the MB solution. S. aureus showed multiple events above the discriminator level (28,000 cps) due to the loop structure of the MB hybridizing to a complementary sequence contained within the genome of S. aureus, whereas the E. coli sample showed no events above this level. The events from the S. aureus predicted a DNA concentration (derived from the calibration curve) of 1.3 × 10−15 M. Based on the DNA copy number sampled during the experiment, more than one copy of the gene was present in each cell. If 20 copies of the gene were present in each cell, the data secured from the calibration curve would be in good agreement to that predicated on the input copy number. Kim et al. demonstrated that the copy number of the 16S ribosomal gene varies from 1 to 15.48
The limit-of-detection for this system is basically a single copy of DNA, indicating that the cell detection limit is 1 cell. However, the ability to detect a single copy depends on the single-molecule sampling efficiency to be near unity and the sampling throughput to be relatively high. The sampling throughput is set by the linear velocity and the input volume, with higher linear velocities increasing the throughput but dropping the single-molecule SNR due to reduced transit times limiting the amount of photons extracted from the molecule.44 Also, larger probe volumes can enhance the sampling throughput, but this can reduce the SNR for the SMD measurements because of larger scattering contributions to the background.
Conclusions
We have demonstrated the ability to detect single molecules using a small footprint instrument coupled to microfluidics that will find applications where point-of-use measurements with near real-time results are required. In addition, the employed optical train consisting of integrated fiber optic cables to the microfluidic for both excitation and emission minimized end user optical alignment, which significantly reduced operator expertise requirements; only insertion of the cable termini into SMA connectors was required to install a new chip. Also, the implementation of a FPGA in conjunction with a diode laser and SPAD significantly reduced the instrument footprint compared to conventional single-molecule optical components and data processing electronics. As a demonstration of the utility of this instrument for analyses using SMD, MBs were designed to probe bacterial cells for the gene encoding the Gram(+) identity using S. aureus and E. coli as models.In the current format, the target cells were isolated, lysed and the genomic DNA purified off-chip. However, we can envision adding additional fluidic sample processing modules to the existing microfluidic to provide the ability to fully prepare the sample prior to performing the single-molecule measurement. For example, we can select cells,49–51 lyse the selected cells either chemically or electrically, isolate the target materials using solid-phase extraction,52–54 and then perform the molecular reactions on the purified material.55–57 This will create a fully integrated system to accept a variety of input samples, for example clinical samples used for in vitro diagnostics, prepare the samples and then perform SMD without requiring significant operator intervention or large amounts of operator expertise.
Acknowledgements
The authors would like to thank the National Institutes of Health (EB-010087), the National Science Foundation (EPS-0346411) and the Louisiana Board of Regents for financial support of this work. The authors would also like to thank the World Class University program (R32-20054) of South Korea (Ulsan National Institute of Science and Technology, UNIST) for partial financial support of this work as well.References
- C. Gell, T. Sabir, J. Westwood, A. Rashid, D. A. M. Smith, S. A. Harris and P. G. Stockley, J. Mol. Biol., 2008, 384, 264–278 CrossRef CAS.
- U. Haupts, M. Rudiger, S. Ashman, S. Turconi, R. Bingham, C. Wharton, J. Hutchinson, C. Carey, K. J. Moore and A. J. Pope, J. Biomol. Screening, 2003, 8, 19–33 CrossRef CAS.
- M. Hintersteiner and M. Auer, in Fluorescence Methods and Applications: Spectroscopy, Imaging, and Probes, ed. O. S. Wolfbeis, 2008, vol. 1130, pp. 1–11 Search PubMed.
- S. H. Kang, Y. J. Kim and E. S. Yeung, Anal. Bioanal. Chem., 2007, 387, 2663–2671 CrossRef CAS.
- K. J. Moore, S. Turconi, S. Ashman, M. Ruediger, U. Haupts, V. Emerick and A. J. Pope, J. Biomol. Screening, 1999, 4, 335–353 CrossRef CAS.
- L. A. Neely, S. Patel, J. Garver, M. Gallo, M. Hackett, S. McLaughlin, M. Nadel, J. Harris, S. Gullans and J. Rooke, Nat. Methods, 2006, 3, 41–46 CrossRef CAS.
- H. Qiu, E. P. Ferrell, N. Nolan, B. H. Phelps, R. Tabibiazar, D. H. Whitney and E. A. Nalefski, Clin. Chem., 2007, 53, 2010–2012 CAS.
- L. Wang, G. Xu, Z. K. Shia, W. Jiang and W. R. Jin, Anal. Chim. Acta, 2007, 590, 104–109 CrossRef CAS.
- A. H. B. Wu, N. Fukushima, R. Puskas, J. Todd and P. Goix, Clin. Chem., 2006, 52, 2157–2159 CAS.
- M. B. Wabuyele, H. Farquar, W. Stryjewski, R. P. Hammer, S. A. Soper, Y. W. Cheng and F. Barany, J. Am. Chem. Soc., 2003, 125, 6937–6945 CrossRef CAS.
- E. B. Shera, N. K. Seitzinger, L. M. Davis, R. A. Keller and S. A. Soper, Chem. Phys. Lett., 1990, 174, 553–557 CrossRef CAS.
- S. A. Soper, E. B. Shera, J. C. Martin, J. H. Jett, J. H. Hahn, H. L. Nutter and R. A. Keller, Anal. Chem., 1991, 63, 432–437 CrossRef CAS.
- D. Erickson, D. Sinton and D. Q. Li, Lab Chip, 2004, 4, 87–90 RSC.
- A. Giudice, M. Ghioni, R. Biasi, F. Zappa, S. Cova, P. Maccagnani and A. Gulinatti, J. Mod. Opt., 2007, 54, 225–237 CrossRef.
- A. Agrawal, C. Y. Zhang, T. Byassee, R. A. Tripp and S. M. Nie, Anal. Chem., 2006, 78, 1061–1070 CrossRef CAS.
- D. L. Yin, D. W. Deamer, H. Schmidt, J. P. Barber and A. R. Hawkins, Opt. Lett., 2006, 31, 2136–2138 CrossRef CAS.
- P. S. Dittrich and A. Manz, Anal. Bioanal. Chem., 2005, 382, 1771–1782 CrossRef CAS.
- F. Yan and T. Vo-Dinh, Sens. Actuators, B, 2007, 121, 61–66 CrossRef.
- S. J. Mechery, X. J. J. Zhao, L. Wang, L. R. Hilliard, A. Munteanu and W. H. Tan, Chem.–Asian J., 2006, 1, 384–390 CrossRef CAS.
- J. C. Fister III, S. C. Jacobson, L. M. Lloyd, M. Davis and J. M. Ramsey, Anal. Chem., 1998, 70, 431–437 CrossRef.
- E. Verpoorte, Lab Chip, 2003, 3, 42N–52N RSC.
- S. Camou, H. Fujita and T. Fujii, Lab Chip, 2003, 3, 40–45 RSC.
- J. Seo and L. P. Lee, Sens. Actuators, B, 2004, 99, 615–622 CrossRef.
- J. C. Roulet, R. Volkel, H. P. Herzig, E. Verpoorte, N. F. de Rooij and R. Dandliker, Anal. Chem., 2002, 74, 3400–3407 CrossRef CAS.
- M. L. Chabinyc, D. T. Chiu, J. C. McDonald, A. D. Stroock, J. F. Christian, A. M. Karger and G. M. Whitesides, Anal. Chem., 2001, 73, 4491–4498 CrossRef CAS.
- A. E. Bruno, F. Maystre, B. Krattiger, P. Nussbaum and E. Gassmann, TrAC, Trends Anal. Chem., 1994, 13, 190–198 CrossRef CAS.
- J. J. Deng and Y. T. Huang, J. Lightwave Technol., 1998, 16, 1062–1069 CrossRef.
- M. Galarza, K. De Mesel, R. Baets, A. Martinez, C. Aramburu and M. Lopez-Amo, Appl. Opt., 2003, 42, 4841–4846 CrossRef CAS.
- M. Galarza, K. De Mesel, S. Verstuyft, U. Aramburu, M. Lopez-Amo, I. Moerman, P. Van Daele and R. Baets, J. Lightwave Technol., 2003, 21, 269–274 CrossRef.
- J. Hubner, K. B. Mogensen, A. M. Jorgensen, P. Friis, P. Telleman and J. P. Kutter, Rev. Sci. Instrum., 2001, 72, 229–233 CrossRef CAS.
- N. M. Litchinitser, A. K. Abeeluck, C. Headley and B. J. Eggleton, Opt. Lett., 2002, 27, 1592–1594 CrossRef CAS.
- K. N. Ren, Q. L. Liang, B. Yao, G. O. Luo, L. D. Wang, Y. Gao, Y. M. Wang and Y. Qiu, Lab Chip, 2007, 7, 1574–1580 RSC.
- Q. Xiang, G. Q. Hu, Y. L. Gao and D. Q. Li, Biosens. Bioelectron., 2006, 21, 2006–2009 CrossRef CAS.
- S. A. Soper, S. M. Ford, Y. C. Xu, S. Z. Qi, S. McWhorter, S. Lassiter, D. Patterson and R. C. Bruch, J. Chromatogr., A, 1999, 853, 107–120 CrossRef CAS.
- U. Dharmasiri, M. A. Witek, A. A. Adams, J. K. Osiri, M. L. Hupert, T. S. Bianchi, D. L. Roelke and S. A. Soper, Anal. Chem., 2010, 82, 2844–2849 CrossRef CAS.
- M. G. Lorenz, L. Mengibar, L. Entrena and R. Sanchez-Reillo, in Field-Programmable Logic and Applications, Proceedings, ed. P. Y. K. Cheung, G. A. Constantinides and J. T. DeSousa, 2003, vol. 2778, pp. 220–229 Search PubMed.
- S. R. Chowdhury, D. Chakrabarti and H. Saha, Microprocessors and Microsystems, 2008, 32, 107–120 CrossRef.
- A. Castro and R. T. Okinaka, Analyst, 2000, 125, 9–11 RSC.
- A. Castro and J. G. K. Williams, Anal. Chem., 1997, 69, 3915–3920 CrossRef CAS.
- Z. Peng, S. A. Soper, M. R. Pingle and F. Barany, Anal. Chem., 2010, 82, 9727–9735 CrossRef CAS.
- M. L. Hupert, W. J. Guy, S. D. Llopis, H. Shadpour, S. Rani, D. E. Nikitopoulos and S. A. Soper, Microfluid. Nanofluid., 2007, 3, 1–11 CrossRef CAS.
- K. K. Ojo, D. Tung, H. Luis, M. Bernardo, J. Leitao and M. C. Roberts, FEMS Microbiology Letters, 2004, 238, 411–416 CAS.
- J. Emory and S. A. Soper, Anal. Chem., 2008, 80, 3897–3903 CrossRef CAS.
- S. A. Soper, Q. L. Mattingly and P. Vegunta, Anal. Chem., 1993, 65, 740–747 CrossRef CAS.
- S. A. Soper, H. L. Nutter, R. A. Keller, L. M. Davis and E. B. Shera, Photochem. Photobiol., 1993, 57, 972–977 CrossRef CAS.
- Z. Peng, S. A. Soper, M. R. Pingle, F. Barany and L. M. Davis, Anal Chem, 82, pp. 9727–9735 Search PubMed.
- N. Panchuk-Voloshina, R. P. Haugland, J. Bishop-Stewart, M. K. Bhalgat, P. J. Millard, F. Mao, W.-Y. Leung and R. P. Haugland, J. Histochem. Cytochem., 1999, 47, 1179–1188 CrossRef CAS.
- J.-s. Kim and N. Wang, BMC Res. Notes, 2009, 2, 37 CrossRef.
- A. A. Adams, P. I. Okagbare, J. Feng, M. L. Hupert, D. Patterson, J. Gottert, R. L. McCarley, D. Nikitopoulos, M. C. Murphy and S. A. Soper, J. Am. Chem. Soc., 2008, 130, 8633–8641 CrossRef CAS.
- U. Dharmasiri, S. Balamurugan, A. A. Adams, P. I. Okagbare, A. Obubuafo and S. A. Soper, Electrophoresis, 2009, 30, 3289–3300 CrossRef CAS.
- U. Dharmasiri, S. Balamurugan, R. L. McCarley, D. Spivak and S. A. Soper, Electrophoresis, 2009, 30, 3289–3300 CrossRef CAS.
- Y. C. Xu, B. Vaidya, A. B. Patel, S. M. Ford, R. L. McCarley and S. A. Soper, Anal. Chem., 2003, 75, 2975–2984 CrossRef CAS.
- M. A. Witek, S. Llopis, A. Wheatley, R. McCarley and S. A. Soper, Nucleic Acids Res., 2006, 34, e74 CrossRef.
- M. A. Witek, M. L. Hupert, D. S. W. Park, K. Fears, M. C. Murphy and S. A. Soper, Anal. Chem., 2008, 80, 3483–3491 CrossRef CAS.
- M. Hashimoto, P. C. Chen, M. W. Mitchell, D. E. Nikitopoulos, S. A. Soper and M. C. Murphy, Lab Chip, 2004, 4, 638–645 RSC.
- J. F. Chen, M. Wabuyele, H. W. Chen, D. Patterson, M. Hupert, H. Shadpour, D. Nikitopoulos and S. A. Soper, Anal. Chem., 2005, 77, 658–666 CrossRef CAS.
- M. Hashimoto, M. L. Hupert, M. C. Murphy, S. A. Soper, Y. W. Cheng and F. Barany, Anal. Chem., 2005, 77, 3243–3255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1an15658f |
| This journal is © The Royal Society of Chemistry 2012 |