Solvent–gelator interactions—using empirical solvent parameters to better understand the self-assembly of gel-phase materials
William
Edwards
,
Cecile A.
Lagadec
and
David K.
Smith
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: dks3@york.ac.uk; Fax: +44 (0)1904434181
First published on 9th October 2010
Abstract
By studying a family of L-lysine bis-urea gelators with variable peripheral groups in different solvents, a more detailed understanding of the way in which gelator fibres interact with the surrounding solvent environment is obtained. In all cases, these gelators establish the same hydrogen bonding molecular recognition pathways—however, this process is mediated by the nature of the solvent. In terms of Kamlet–Taft parameters, the α parameter of the solvent (hydrogen bond donor ability) has primary importance in controlling whether the gelator can establish a hydrogen bond network; the β parameter (hydrogen bond acceptor ability) plays a secondary role in tuning the thermal stability of the gel, and the π* parameter (polarisability) controls the solvation of the peripheral groups on the gelator by the solvent, and hence tunes the gel stability and the ability of the gelator to establish fibre–fibre interactions, as evidenced by scanning electron microscopy imaging. Considering solvent parameters allows us to gain a unique insight into hierarchical assembly processes at different length scales, i.e., molecular scale gelator–gelator interactions, and nanoscale fibre–fibre and fibre–solvent interactions. These processes are at the heart of developing effective models for the dynamic assembly of gel-phase soft materials.
Introduction
Supramolecular gels, constructed by the self-assembly of molecular-scale building blocks, constitute a key class of soft nanostructured material.1 Such materials are colloidal in nature, with the low molecular weight building blocks assembling into a supramolecular polymer scaffold which acts as a three-dimensional ‘solid-like’ network to immobilise the flow of the bulk ‘liquid-like’ solvent phase in which it is formed. Such materials are widely applied in an industrial setting as lubricants and viscosity modifiers,2 and high-tech applications of such materials in fields as diverse as tissue engineering and nanoscale electronics are currently being proposed and tested.3The majority of papers in the field focus on the influence of gelator structure on assembly, and largely ignore the role of solvent—perhaps surprising given that the ‘liquid-like’ phase typically constitutes ca. 99% of these materials by weight. Research sometimes attempts to correlate gel behaviour with the nature of the solvent in a qualitative manner,4 but very few studies attempt to gain a quantitative insight into the precise ways in which solvent can influence self-assembly and gelation. Gaining this type of fundamental understanding is essential if the behaviour of this important class of self-assembled soft material is to be truly understood.5
There are a range of different ways in which the behaviour of solvents can be parameterised and understood.6 The simplest approach to quantifying solvent effects is to employ bulk solvent parameters, such as dielectric constant. This has been done in an attempt to correlate the thermal behaviour of gels.7 However, it is well known that bulk solvent parameters can reflect only very roughly the interactions between a solute and the solvent. For this reason, a range of different solvent scales have been developed and many of these have been applied to gel-phase materials. In early work, Hanabusa and co-workers reported that the minimum gelation concentration of cyclo(dipeptide) gelators was dependent on Hildebrand's solvent polar solubility parameter.8 We also applied a similar understanding to the assembly of gelators based on dendritic peptide building blocks,9 an approach which was subsequently also used by Zhu and Dordick to predict hydrogen bond mediated gelation.10 In related work, other researchers have employed Reichardt's ET(30) parameter scale, and the Hansen hydrogen bonding solubility parameter index, to correlate gel behaviour.11 Boutellier and co-workers have noted that the steric demands of solvent molecules can be parameterised and shown to have a direct influence on gel formation.12
An alternative, and more detailed way of accounting for specific interactions between solvent and solute, is to apply the Kamlet–Taft parameters.13 There are three different Kamlet–Taft parameters, each of which accounts for a different way in which a solvent can interact with a solute: α (hydrogen bond donor ability), β (hydrogen bond acceptor ability) and π* (dipolarity/polarisability). In spite of their known importance in physical organic chemistry, these parameters have not been widely used to consider supramolecular gelation. Aggeli and co-workers demonstrated that only by combining the dielectric constant of the solvent with its hydrogen bond acceptor ability, α, could they gain an effective insight into solvent effects on gelation.14 We have also noted that Kamlet–Taft parameters, in particular α, can have a degree of predictive power for gelation processes,9,15 but have not previously uncovered any detailed relationships.
In an attempt to better understand these essential solvent effects on gelation, this paper reports the investigation of a high-throughput library of bis-urea gelators based on an amino acid, L-lysine, framework. Bis-urea gelators are well-known in the literature—they assemble as a consequence of complementary urea–urea interactions (Fig. 1).16 They are widely used as thickening agents in industrial applications—as such, they constitute an important class of applied material.2Lysine-derived bis-ureas are synthesised by the simple one-pot reaction between L-lysine methyl ester diisocyanate and amines, as previously reported by Hanabusa and co-workers (Scheme 1).17 Such gelators are highly versatile as they contain two urea hydrogen bonding units, a chiral centre and a functionalisable ‘handle’ (methyl ester). Bis-ureas are robust gelators, with the urea–urea hydrogen bond interactions being maintained in the presence of a wide range of different peripheral R groups. For example, by increasing the hydrophilicity of the peripheral groups, it is possible for bis-urea systems to act as hydrogelators in water.18 We have also recently demonstrated that the introduction of an allyl group onto the lysine bis-urea framework leads to a simple, yet versatile gelator which can assemble into either fibres or fibrillar vesicles, and in which the alkenes can be subsequently cross-linked by metathesis.19
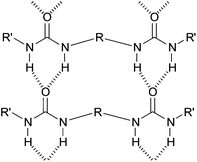 | ||
| Fig. 1 Schematic diagram of general mode of assembly of bis-ureas. | ||
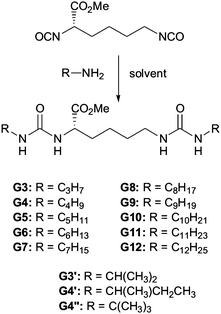 | ||
| Scheme 1 Schematic diagram of general mode of assembly of bis-ureas. | ||
In this new study, we hoped that by subtly modifying the peripheral groups of the gelator, we would gain a more detailed insight into the way in which solvent effects mediate gel assembly.
Results and discussion
Synthesis of gelators
The gelators were synthesised via simple reaction between L-lysine methyl ester diisocyanate and the appropriate amine to form the bis-urea. The reactions were carried out in parallel on a carousel reactor with multiple different amines in order to generate a small library of potential gelators (Scheme 1). Some of these compounds (G6, G8 and G12) have been previously reported in the literature, although the gels were previously investigated in situ, directly after reaction of the two components.17 In this case, however, the product gelators were purified by recrystallisation from methanol/ether and pure bis-ureas were produced in yields of 43–91%. Two families of molecules were synthesized—the first containing a range of linear aliphatic R groups (from C3H7 to C12H25) and the second containing several isomeric R groups with branched structures (Scheme 1).Thermal stabilities of gels
The bis-ureas were screened for gelation ability in nine carefully selected organic solvents—toluene (TOL), benzene (BZ), 1,2-dichlorobenzene (DCB), 1,2-dimethoxyethane (DME), ethyl acetate (EtOAc), acetonitrile (ACN), dichloromethane (DCM), chloroform (CHCl3) and methanol (MeOH). These solvents were chosen because they exhibit a range of different characteristics in terms of hydrogen bonding (acceptor/donor) ability and polarity.In each case, the samples used for screening were made by weighing 15 mg of gelator into a sample tube, adding 0.5 ml of solvent, sonicating for 30 min and heating the sample to just below the boiling point of the solvent. All samples were then left to stand for 24 h prior to recording their thermal behaviour. The thermal stability of the gels was estimated by measuring the Tgel values of the different samples (the temperature at which the gel is converted into a sol). This was achieved by reproducible tube inversion methodology,20 with the sample being heated at 0.5 °C min−1. When a drop of solvent ran from the inverted gel sample, the temperature was recorded as the Tgel. The data are presented in Table 1. All gelators were investigated at a relatively high mass loading of 3% w/v—ensuring that all compounds capable of forming gels were in the ‘plateau region’21 for which gelation is independent of concentration.†
| TOL | BZ | DCB | DME | EtOAc | |
|---|---|---|---|---|---|
| a TOL = toluene, BZ = benzene, DCB = 1,2-dichlorobenzene, DME = 1,2-dimethoxyethane, and EtOAc = ethyl acetate. | |||||
| G3 | PG | PG | OG (27 °C) | PG | PG |
| G4 | PG | PG | OG (25 °C) | Ins | PG |
| G5 | PG | PG | OG (25 °C) | Ins | PG |
| G6 | PG | CG (55 °C) | OG (25 °C) | Ins | OG (5 °C) |
| G7 | CG (70 °C) | CG (60 °C) | OG (28 °C) | OG (7 °C) | OG (12 °C) |
| G8 | CG (73 °C) | CG (60 °C) | OG (28 °C) | OG (4 °C) | OG (10 °C) |
| G9 | CG (73 °C) | CG (62 °C) | OG (38 °C) | OG (13 °C) | OG (12 °C) |
| G10 | CG (74 °C) | CG (63 °C) | OG (46 °C) | OG (14 °C) | OG (12 °C) |
| G11 | CG (74 °C) | CG (67 °C) | OG (50 °C) | OG (16 °C) | OG (14 °C) |
| G12 | CG (73 °C) | CG (70 °C) | OG (49 °C) | OG (14 °C) | OG (15 °C) |
Table 1 presents the solvents which were most supportive of gelation, while other solvents investigated were less supportive of gelation. In particular, the use of acetonitrile (ACN) only led to the formation of weak gels, and Tgel values could not be determined, as they are below ca. 0 °C, which is difficult to accurately determine on experimental grounds. Dichloromethane is highly volatile, and solvent evaporation was somewhat problematic during gel preparation—however, again, only partial gels could be formed at best. In chloroform, no gelation was observed. It was also noted that gelation was not observed in protic organic solvents such as methanol.
Application of Kamlet–Taft parameters
In order to understand the solvent effects on gelation, we decided to consider the Kamlet–Taft parameters for the different solvents (Table 2).13 We anticipated that using these parameters may allow us to break down the differential effects of solvent on gelation.| Solvent a | α | β | π* |
|---|---|---|---|
| a TOL = toluene, BZ = benzene, DCB = 1,2-dichlorobenzene, DME = 1,2-dimethoxyethane, EtOAc = ethyl acetate, ACN = acetonitrile, DCM = dichloromethane, CHCl3 = chloroform, and MeOH = methanol. | |||
| TOL | 0.00 | 0.11 | 0.49 |
| BZ | 0.00 | 0.10 | 0.55 |
| DCB | 0.00 | 0.03 | 0.77 |
| DME | 0.00 | 0.41 | 0.53 |
| EtOAc | 0.00 | 0.45 | 0.45 |
| ACN | 0.19 | 0.31 | 0.66 |
| DCM | 0.30 | 0.00 | 0.73 |
| CHCl3 | 0.44 | 0.00 | 0.69 |
| MeOH | 0.93 | 0.62 | 0.60 |
From consideration of Tables 1 and 2, it is evident that gels are best supported in those solvents where the α parameter is zero—i.e., TOL, BZ, DCB, DME and EtOAc. None of these solvents are able to donate hydrogen bonds to the gelator (the solute). In comparison hydrogen bond donor solvents, with relatively high α values, such as CHCl3 and MeOH, do not support gelation at all, instead giving rise to clear solutions. Furthermore, ACN and DCM which have intermediate α values of 0.19 and 0.30 can only support partial unstable gels. These observations are in agreement with previous studies which have demonstrated that the α parameter has the major predictive power over whether a hydrogen bonding gelator will self-assemble in a given solvent.9,14,15,22 Clearly if the solvent is a good hydrogen bond donor, it can donate hydrogen bonds to the gelator, with these competitive interactions preventing the establishment of gelator–gelator hydrogen bonding networks.
On first inspection of the data, the β and π* parameters appear to be less important—for example, DCB and EtOAc have low and high β values, respectively, while TOL and DCB have low and high π* values, respectively, but in all cases, gelation can still occur. However, on examining the data in more detail, it becomes clear that the most thermally stable gels are formed in TOL and BZ—both of which have low β and π* values. Gels in DCB, which has a high π* value are intermediate in thermal stability, whilst gels formed in DME and EtOAc, both of which have relatively high β values, have the least thermal stability. We would therefore conclude that of the other parameters, if the β value is too high, it depresses thermal stability (Tgel). The β parameter represents the ability of the solvent to accept hydrogen bonds—for example from the N–H groups of the gelator—and this clearly disrupts self-assembly and thermal stability to some extent. The π* (polarisability) parameter then plays a smaller role in controlling thermal stability.
In combination, these observations can be summarised as follows. Hydrogen bond mediated gelation (i) is primarily dependent on α (hydrogen bond donor ability of solvent), (ii) has a secondary dependence on β (hydrogen bond acceptor ability of solvent), and (iii) exhibits a small dependence on π* (polarisability of solvent).
We then went on to consider in detail the way in which the alkyl chains on the periphery of the gelator modified the effect of solvent on gelation. It might be expected that these peripheral functional groups are the part of the gelator structure in direct contact with the solvent, and that they should therefore play a role in controlling the interaction of the gelator with different solvents. However, such effects have not previously been quantified. We therefore reasoned that this might provide us with a more detailed insight into the gelation process. In order to achieve this, we studied the gels formed in the solvents for which α = 0.00, in order to determine the combined effect of chain length and the other solvent parameters on gelation (Fig. 2).
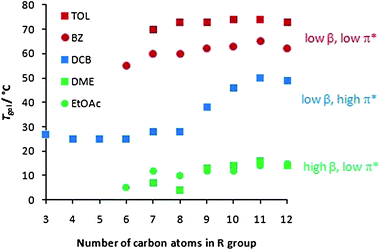 | ||
| Fig. 2 Effect of alkyl chain length on gel–sol transition temperature (Tgel) in toluene (TOL), benzene (BZ), 1,2-dichlorobenzene (DCB), 1,2-dimethoxyethane (DME) and ethyl acetate (EtOAc). | ||
Firstly, the solvents can clearly be divided into three general types—as explained above. Those solvents with low β and π* values (TOL and BZ), which do not compete with gelator–gelator interactions, form the most thermally stable gels. Those solvents with elevated β values (DME and EtOAc) can compete with the hydrogen bonding pathways within the gel, and hence form the gels with the lowest thermal stabilities. Finally, DCB has an elevated π* value which depresses gelation to some extent, meaning these gels have intermediate thermal stabilities.
Looking at the data in more detail, it can be seen that in DCB, all chain lengths support gelation to some extent, and there is a general increase in thermal stability with increasing chain length. However, in the other solvents, a critical chain length is required for gelation to be observed, but as the chain length extends further, there are no additional benefits on thermal stability. It has previously been suggested that the effect of lengthening alkyl chains can be best rationalised in terms of increasing van der Waals interactions.17a,b,23 However, it is clear from Fig. 2 that van der Waals interactions may not provide a complete explanation of the impact of alkyl chains on gelation.
Comparing TOL (or BZ) and DCB, they have similar α and β values, but significantly different π* parameters, with DCB (0.77) being significantly more polar than TOL (0.49). The change in π* therefore has a clear effect on gelation. Attaching long alkyl chains to the gelator makes it relatively apolar (indeed hexane has a π* value of 0.00), and as such, long alkyl chains will be more effectively solvated in apolar TOL than polar DCB, and the gelator is therefore less compatible with DCB than TOL. In DCB, as the aliphatic chain increases in length, so does the driving force for the gelator to aggregate with itself, as it becomes less and less compatible with the relatively polar solvent—in this way, in DCB, increasing the alkyl chain length significantly aids the formation of ‘solid-like’ fibres. In the less polar solvents, TOL and BZ, the long aliphatic chains of the gelator will have to be desolvated in order for self-assembly to take place, and the benefits of any additional van der Waals forces between longer chains are less evident. Furthermore, we would propose that the lower compatibility between DCB and longer alkyl chains encourages a greater degree of gel fibre aggregation and bundling, as the peripheral aliphatic chains are better able to interact with one another than the surrounding solvent. Conversely, we suggest that a critical chain length is required for full gelation in solvents, such as TOL, with low π* values because the shorter alkyl chain lengths limit gelator fibre aggregation and full gel network formation. Supporting evidence for these ideas come from the observation that in DCB, the gelators assemble to give opaque gels (indicative of larger nano/microscale structures), whilst in TOL and BZ, they assemble to give clear gels (indicative of smaller nanoscale structures). SEM evidence is also presented below. We then went on to consider the thermal behaviour of the different gels in DME and EtOAc (Fig. 2). These gels have similar π* parameters to TOL and BZ, and similarly to those solvents, no significant effect of chain length on gel thermal stability was observed.
In a recent paper we argued that modifying the protecting groups on the periphery of a gelator could have a profound effect on gel thermal stability, primarily by mediating the solubility of the gelator.21 In this case, where we have gelators with an apolar periphery, we are arguing that the π* parameter of the solvent helps control the solubility of different gelators and hence mediates the thermal stability of the gel. These observations are in agreement with recent work from Tritt-Goc and co-workers,11a who reported that solvent polarity could have an impact on the stability of hydrogen bonded gels, and that hydrogen bonding effects alone were insufficient to explain the gelation process.
In summary, we therefore propose that for this class of gelator, the ability of a solvent to support self-assembly and gelation is dependent on (in order of importance):
(i) The ability of the solvent to donate hydrogen bonds (α value) which directly disrupts urea–urea interactions. The α value should ideally be zero for effective gelation to be observed.
(ii) The ability of the solvent to accept hydrogen bonds (β parameter) which can disrupt hydrogen bond interactions to some extent and depress the thermal stability of the gels. The lower the β value, the greater the thermal stability (Tgel values) of the gels.
(iii) The ability of the solvent to solvate the apolar peripheral groups of the gelator (π* value) tunes the stability of the gel. In particular, by modulating the solubility of the gelator surface groups, the π* value is able to enhance the effect of the peripheral groups on self-assembly and gelation.
As such, we suggest that α and β parameters modulate the gelator–gelator interactions on the molecular level, whilst the π* parameter has an impact on the nanoscale fibre–fibre and fibre–solvent interactions.24 In this way, considering solvent effects allows us to effectively deconvolute levels of the hierarchical assembly process on different length scales.
Study of isomeric gelators
We then investigated the gelation behaviour of the isomeric gelators, G3′, G4′ and G4″. In general terms, these compounds behaved similarly to gelators G3 and G4 in that they formed the best gels in solvents with α values equal to zero. Branched gelator G3′ appeared to be marginally more effective than its linear isomer G3—this observation is similar to results previously published by Suzuki and co-workers for a related family of gelators.25 Interestingly, compound G4″ was unable to form stable gels, even in the most supportive solvent, instead giving rise to a partial unstable gel. We propose that the steric hindrance of the tert-butyl group limits the packing of the molecular building blocks into a one-dimensional fibrillar assembly.Circular dichroism spectroscopy
In order to confirm our model of gelation, in which the bis-ureas stack to form one-dimensional fibrils (Fig. 1), irrespective of the nature of the peripheral groups, we employed circular dichroism (CD) spectroscopy to gain an insight into the nanoscale chiral organisation of the self-assembled architectures.26 We had to carry out the CD studies in a solvent which was transparent at the absorption wavelength of the bis-urea gelator, and for this reason DME was selected as the solvent of choice.We investigated gelators G8 and G11, which form gels in this solvent. These spectra had maxima at ca. 215 nm, caused by the absorbance of the urea groups, which are placed in a nanoscale chiral environment within the self-assembled fibril (Fig. 3). Both gelators showed almost identical CD spectra, indicating that the mode of self-assembly of both gelators must be the same under these conditions. This supports the viewpoint that such gelators assemble via the formation of hydrogen bonding molecular recognition pathways between the urea groups, with the nature of the peripheral R groups having minimal effect on the internal hydrogen bonding chiral nanoscale organisation of the fibrils. We can therefore confirm that the R groups appear to modify the gelation ability not by changing the mode of self-assembly, but instead by mediating the gelator–gelator, fibre–fibre and fibre–solvent non-covalent interactions.
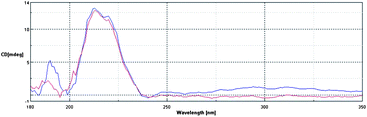 | ||
| Fig. 3 CD spectra of compounds G8 and G11 measured in DME at concentrations of 0.07% w/v. | ||
SEM imaging of gel networks
In order to shed further light on the assembly process, and to probe the nanostructures assembled by these gelators, we performed scanning electron microscopy (SEM). Samples were prepared by leaving a small sample of the gel to dry under ambient conditions on a metal stub. The resulting xerogel was then imaged. Although drying effects can modify observed morphologies, such an approach is particularly useful for providing a comparative view of the behaviour of structurally similar gelators, where differences on drying are assumed to be largely insignificant.Fig. 4 shows the SEM images for gelators G3, G3′, G7 and G11, dried from DCB. All of the images show the presence of fibrillar networks. We have previously reported structurally related lysine-derived gelators in which only the hydrogen bonding units were changed—in that case, the SEM images indicated dramatically different morphologies.27 However, in this case, it appears that varying the peripheral groups has a relatively limited effect on the supramolecular structures which are assembled. Once again, this agrees with our model, in which urea–urea hydrogen bonding interactions give rise to the self-assembly process and are essentially the same for each gelator in the series.
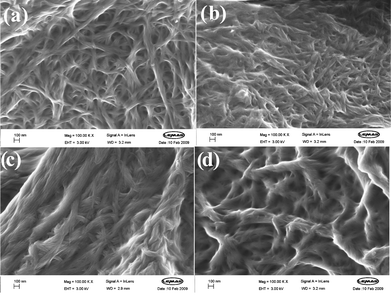 | ||
| Fig. 4 Scanning electron microscopy images of xerogels dried from DCB. (a) G3, (b) G3′, (c) G7, and (d) G11. A higher degree of fibre aggregation is evident for G7 and G11 than for G3 and G3′. In each image the scale bar represents 100 nm. | ||
There are, however, some subtle differences between the images. The images of G3 and G3′ both have a similar appearance, with fibres of roughly the same width (20–30 nm), concurrent with their similar thermal behaviour. Comparing G3 with G7 and G11, however, indicates that although the morphologies are in each case made up from smaller fibres (diameters = ca. 20–30 nm), for G7 and G11 these fibres noticeably aggregate into much larger structures (diameters ca. 150 nm). This bundling process may be a result of a greater degree of fibre–fibre interaction within these gels. As discussed above, in DCB, which is a relatively polar solvent, as the aliphatic chain length increases, there will be a greater driving force towards fibre–fibre interactions, because the gelators have a preference for interacting with one another rather than the solvent. We therefore tentatively propose that the SEM images provide support for a nanoscale change, mediated by the fibre–solvent interaction, and that this change is responsible for the macroscopic observations of changing Tgel behaviour.
Conclusions
In conclusion, by studying a family of simple, structurally related gelators, with variable peripheral groups, we found that in different solvents, the peripheral groups have different effects on gelation. In this way, we have gained a more detailed understanding of the way in which gelator fibres can interact with their solvent environment. Importantly, we have considered the effect of solvent in terms of Kamlet–Taft parameters:• the α parameter (hydrogen bond donor ability) appears to have primary importance in controlling whether a hydrogen bond network of gelators can be established,
• the β parameter (hydrogen bond acceptor ability) appears to tune the thermal stability of the gel by modulating gelator–gelator interactions, and
• the π* parameter (polarisability) appears to control the interactions of the peripheral groups on the gelator with solvent, and tunes gel stability and the ability of the gelator to establish fibre–fibre interactions.
Understanding gelator–gelator, fibre–fibre and fibre–solvent interactions is at the heart of developing effective models for the dynamic assembly of these intriguing colloidal nanostructured materials, and as such, we are currently continuing to develop our fundamental understanding of different classes of gelator in a range of different solvent environments.
Experimental
Materials and methods
Chemical and general reagents were commercially available and used without further purification unless otherwise stated. 1H NMR and 13C NMR were recorded on JEOL ECX400 (1H 400 MHz, 13C 100 MHz) spectrometer. The chemical shifts (δ) are given in units ppm relative to solvent reference. Coupling constants (J) are reported in hertz. Multiplicity for 1H is shown as s (singlet), d (doublet), t (triplet), m (multiplet) or a combination of these. Deuterated chloroform (CDCl3), methanol (CD3OD) and dimethyl sulfoxide (DMSO-d6) solutions were used to record NMR spectra of the compounds. Electrospray ionisation MS were recorded on a Bruker Micro-TOF mass spectrometer. Melting points (mp) were determined using an Olympius BX50 microscope coupled with an heating stage; samples were heated until completely converted to a liquid phase then cooled down to the solid state again. The melting points were determined by heating slowly the sample until the sol–liq transition temperature was observed. Infrared spectra were recorded using a Shimadzu IR Prestige 2 FTIR. Bands from the IR spectra are reported with peak intensities (s, m, and w; strong, medium, and weak). The SEM was carried out on a LEO 1530 Gemini FEG-SEM, images were provided by John P. Harrington at LEMAS (University of Leeds). Gelators G6, G8 and G12 have been previously reported,16 however, they were made in situ and therefore full analytical data are reproduced here for completeness.Procedure for making gels
An accurately measured mass of gelator was weighed out into a 2 ml glass vial. The solvent (0.5 ml) was then added using a Gilson pipette. The sample was sonicated for 30 minutes and let to rest before being heated with a heat gun until a homogeneous solution had been obtained, then was left to cool down. All gels were left overnight to set.Procedure for the measurement of Tgel values
Once the gel was formed it was placed into a high precision thermoregulated oil bath and the heat was increased at the rate of 0.5 °C min−1. The temperature at which solvent ran from the sample was recorded as the Tgel value, which reflects the temperature of the gel–sol transition.Procedure for the preparation of SEM samples
A small amount of gel sample was removed from its glass vial with a spatula and it was spread thinly onto a metal stub (SEM) and left to dry overnight under a fume hood. Before imaging the sample was covered in a thin layer (4 nm) of Pd/Pt busing an Agar sputter coater before being placed in the microscope.General procedure for synthesis of gelators
Gelators were synthesised by adding the appropriate amine (4.96 mmol, 2.1 equiv.) dropwise to a solution of methyl 2,6-diisocyanatohexanoate (lysine diisocyanate methyl ester, LDI) (0.50 g, 2.36 mmol, 1 equiv.) in toluene (20 ml) at 0 °C. The mixture was then heated to 80 °C and stirred for 24 h. The toluene was removed by rotary evaporation. The product was recrystallised from methanol/diethyl ether and dried in a vacuum oven for 24 h.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, ester), 1627s (νCO, urea), 1566s (δN–H, urea), 1258m, 648m. ESI-MS (m/z) C15H32N4O4 [M + H]+ requires 331.2340; found: 331.2339 (100%, [M + H]+), 353.2154 (56%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 640m. ESI-MS (m/z) C15H31N4O4 [M + H]+ requires 331.2340; found: 331.2349 (100%, [M + H]+), 353.2160 (49%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1567s (δN–H, urea), 1258m, 640m. ESI-MS (m/z) C17H35N4O4 [M + H]+ requires 359.2653; found: 359.2657 (100%, [M + H]+), 381.2470 (49%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1219m, 648m. ESI-MS (m/z) C17H35N4O4 [M + H]+ requires 359.2653; found: 359.2656 (100%, [M + H]+), 381.2472 (81%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1550s (δN–H, urea), 1211m, 640m. ESI-MS (m/z) C17H35N4O4 [M + H]+ requires 359.2653; found: 359.2652 (100%, [M + H]+), 381.2468 (93%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 640m. ESI-MS (m/z) C19H39N4O4 [M + H]+ requires 387.2966; found: 387.2966 (100%, [M + H]+), 409.2786 (41%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1219m, 632m. ESI-MS (m/z) C21H43N4O4 [M + H]+ requires 415.3279; found: 415.3279 (100%, [M + H]+), 437.3091 (32%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 633m. ESI-MS (m/z) C23H47N4O4 [M + H]+ requires 443.3592; found: 443.3592 (100%, [M + H]+), 465.3404 (33%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 633m. ESI-MS (m/z) C25H51N4O4 [M + H]+ requires 471.3195; found: 471.3911 (100%, [M + H]+), 493.3727 (46%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1265m, 656m. ESI-MS (m/z) C27H55N4O4 [M + H]+ requires 499.4207; found: 499.4207 (100%, [M + H]+), 521.4022 (56%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1258m, 623m. ESI-MS (m/z) C29H59N4O4 [M + H]+ requires 527.4535; found: 527.4532 (100%, [M + H]+), 549.4343 (25%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1257m, 623m. ESI-MS (m/z) C31H63N4O4 [M + H]+ requires 555.4844; found: 555.4830 (100%, [M + H]+), 577.4635 (30%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1258m, 623m. ESI-MS (m/z) C31H62N4O4 [M] requires 583.5157; (ES+) found: 583.5145 (100%, [M + H]+).
O, ester), 1627s (νCO, urea), 1566s (δN–H, urea), 1258m, 648m. ESI-MS (m/z) C15H32N4O4 [M + H]+ requires 331.2340; found: 331.2339 (100%, [M + H]+), 353.2154 (56%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 640m. ESI-MS (m/z) C15H31N4O4 [M + H]+ requires 331.2340; found: 331.2349 (100%, [M + H]+), 353.2160 (49%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1567s (δN–H, urea), 1258m, 640m. ESI-MS (m/z) C17H35N4O4 [M + H]+ requires 359.2653; found: 359.2657 (100%, [M + H]+), 381.2470 (49%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1219m, 648m. ESI-MS (m/z) C17H35N4O4 [M + H]+ requires 359.2653; found: 359.2656 (100%, [M + H]+), 381.2472 (81%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1550s (δN–H, urea), 1211m, 640m. ESI-MS (m/z) C17H35N4O4 [M + H]+ requires 359.2653; found: 359.2652 (100%, [M + H]+), 381.2468 (93%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 640m. ESI-MS (m/z) C19H39N4O4 [M + H]+ requires 387.2966; found: 387.2966 (100%, [M + H]+), 409.2786 (41%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1219m, 632m. ESI-MS (m/z) C21H43N4O4 [M + H]+ requires 415.3279; found: 415.3279 (100%, [M + H]+), 437.3091 (32%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 633m. ESI-MS (m/z) C23H47N4O4 [M + H]+ requires 443.3592; found: 443.3592 (100%, [M + H]+), 465.3404 (33%, [M + Na]+).
O, ester), 1628s (νCO, urea), 1566s (δN–H, urea), 1258m, 633m. ESI-MS (m/z) C25H51N4O4 [M + H]+ requires 471.3195; found: 471.3911 (100%, [M + H]+), 493.3727 (46%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1265m, 656m. ESI-MS (m/z) C27H55N4O4 [M + H]+ requires 499.4207; found: 499.4207 (100%, [M + H]+), 521.4022 (56%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1258m, 623m. ESI-MS (m/z) C29H59N4O4 [M + H]+ requires 527.4535; found: 527.4532 (100%, [M + H]+), 549.4343 (25%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1257m, 623m. ESI-MS (m/z) C31H63N4O4 [M + H]+ requires 555.4844; found: 555.4830 (100%, [M + H]+), 577.4635 (30%, [M + Na]+).
O, ester), 1620s (νCO, urea), 1566s (δN–H, urea), 1258m, 623m. ESI-MS (m/z) C31H62N4O4 [M] requires 583.5157; (ES+) found: 583.5145 (100%, [M + H]+).
Acknowledgements
We thank EPSRC and Givaudan for funding, and acknowledge John Harrington (University of Leeds) for SEM.References
- (a) Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. P. Terech and R. G. Weiss, Springer, Dordrecht, The Netherlands, 2006 Search PubMed; (b) D. K. Smith, in Molecular Gels—Nanostructured Soft Materials in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS.
- C. J. Donahue, J. Chem. Educ., 2006, 83, 862–869 CrossRef CAS.
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- For selected recent examples see: (a) S. Bhattacharya and A. Pal, J. Phys. Chem. B, 2008, 112, 4918–4927 CrossRef CAS; (b) P. Xue, R. Lu, X. Yang, L. Zhao, D. Xu, Y. Liu, H. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem.–Eur. J., 2009, 15, 9824–9835 CrossRef CAS; (c) J. Puigmarti-Luis, A. P. del Pino, V. Laukhin, L. N. Feldborg, C. Rovira, E. Laukhina and D. B. Amabilino, J. Mater. Chem., 2010, 20, 466–474 RSC.
- J. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS.
- (a) Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC; (b) C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weiheim, 3rd edn, 2003 Search PubMed.
- J. Makarevic, M. Jokic, M. Peric, V. Tomisic, B. Kojic-Prodic and M. Zinic, Chem.–Eur. J., 2001, 7, 3328–3341 CrossRef CAS.
- K. Hanabusa, M. Matsumoto, M. Kimura, A. Kakehi and H. Shirai, J. Colloid Interface Sci., 2000, 224, 231–244 CrossRef CAS.
- A. R. Hirst and D. K. Smith, Langmuir, 2004, 20, 10851–10857 CrossRef CAS.
- G. Zhu and J. S. Dordick, Chem. Mater., 2005, 18, 5988–5995.
- (a) M. Bielejewski, A. Lapinski, R. Luboradzki and J. Tritt-Goc, Langmuir, 2009, 25, 8274–8279 CrossRef CAS; (b) M. A. Rogers and A. G. Marangoni, Langmuir, 2009, 25, 8556–8566 CrossRef CAS.
- T. Pinault, B. Isare and L. Boutellier, ChemPhysChem, 2006, 7, 816–819 CrossRef CAS.
- (a) M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS; (b) C. Laurence, P. Nicolet, M. T. Dalati, J. L. M. Abboud and R. Notario, J. Phys. Chem., 1994, 98, 5807–5816 CrossRef CAS; (c) Y. J. Marcus, J. Solution Chem., 1991, 20, 929–944 CAS.
- A. Aggeli, N. Bell, J. N. Boden, P. F. Keen, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS.
- K. S. Partridge, D. K. Smith, G. M. Dykes and P. T. McGrail, Chem. Commun., 2001, 319–320 RSC.
- For a review see: (a) F. Fages, F. Vögtle and M. Zinic, Top. Curr. Chem., 2005, 256, 77–131 CASFor key examples see: (b) J. van Esch, S. de Feyter, R. M. Kellogg, F. de Schryver and B. L. Feringa, Chem.–Eur. J., 1997, 3, 1238–1243 CrossRef CAS; (c) F. S. Schoonbeek, J. van Esch, B. Wegewijs, D. B. A. Rep, M. P. de Haas, T. M. Klapwijk, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 1999, 38, 1393–1397 CrossRef CAS; (d) M. George, G. Tan, V. T. John and R. G. Weiss, Chem.–Eur. J., 2005, 11, 3243–3254 CrossRef CAS.
- (a) M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Langmuir, 2003, 19, 8622–8624 CrossRef CAS; (b) M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Org. Biomol. Chem., 2004, 2, 1155–1159 RSC; (c) M. Suzuki, Y. Nakjima, T. Sato, H. Shirai and K. Hanabusa, Chem. Commun., 2006, 377–379 RSC; (d) M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2009, 38, 967–975 RSC.
- L. A. Estroff and A. D. Hamilton, Angew. Chem., Int. Ed., 2000, 39, 3447–3450 CrossRef CAS.
- I. A. Coates and D. K. Smith, Chem.–Eur. J., 2009, 15, 6340–6344 CrossRef CAS.
- S. R. Raghavan and B. H. Cipriano, Gel Formation: Phase Diagrams using Tabletop Rheology and Calorimetry, in Molecular Gels, Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, Netherlands, 2006, ch. 8 Search PubMed.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS.
- C. Lagadec and D. K. Smith, Chem. Commun., 2010 10.1039/c0cc01449d.
- N. Zweep, A. Hopkinson, A. Meetsma, W. R. Browne, B. L. Feringa and J. H. van Esch, Langmuir, 2009, 25, 8802–8809 CrossRef CAS.
- For an example in which changing the solvent has induced microstructural changes see: P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem.–Eur. J., 2010, 16, 3176–3183 Search PubMed.
- M. Suzuki, M. Yumoto, H. Shirai and K. Hanabusa, Tetrahedron, 2008, 64, 10395–10400 CrossRef CAS.
- (a) Circular Dichroism: Principles and Applications, ed. N. Berova, K. Nakanishi and W. W. Woody, Wiley-VCH, New York, 2000 Search PubMed; (b) D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC.
- J. G. Hardy, A. R. Hirst, I. Ashworth, C. Brennan and D. K. Smith, Tetrahedron, 2007, 63, 7397–7406 CrossRef CAS.
Footnote |
| † It should be noted that the molar concentration of each gelator is different. The compounds with longer alkyl chains have higher RMMs, and hence lower concentrations. However, this factor does not influence the thermal stability, as all gelators are being investigated in the concentration regime in which thermal behaviour is independent of concentration. |
| This journal is © The Royal Society of Chemistry 2011 |