DOI:
10.1039/C0SM00618A
(Paper)
Soft Matter, 2011,
7, 237-246
Hydrogel-supported protein-tethered bilayer lipid membranes: a new approach toward polymer-supported lipid membranes
Received
2nd July 2010
, Accepted 1st September 2010
First published on 6th October 2010
Abstract
Polymer-supported bilayer lipid membranes offer great opportunities for the investigation of functional membrane proteins. Here we present a new approach in this direction by introducing a thin hydrogel layer as a soft ‘cushion’ on indium–tin oxide (ITO), providing a smooth, functional surface to form the protein-tethered BLM (ptBLM). ITO was used as a transparent electrode, enabling simultaneous implementation of electrochemical and optical waveguide techniques. The hydrogel poly(N-(2-hydroxyethyl)acrylamide-co-5-acrylamido-1-carboxypentyl-iminodiacetate-co-4-benzoylphenyl methacrylate) (P(HEAAm-co-NTAAAm-co-MABP)) was functionalized with the nickel chelating nitrilotriacetic acid (NTA) groups, to which cytochrome c oxidase (CcO) from Paracoccus denitrificans was bound in a well defined orientation via a his-tag attached to its subunit I. Given that the mesh size of P(HEAAm-co-NTAAAm-co-MABP) was smaller than the protein size, binding to the hydrogel occurred only on the top of the layer. The lipid bilayer was formed around the protein by in situdialysis. Electrochemical impedance spectroscopy showed good electrical sealing properties with a resistance of ∼1 MΩ cm2. Furthermore, surface plasmon resonance optical waveguide spectroscopy (SPR/OWS) indicated an increased anisotropy of the system after formation of the lipid bilayer. Cyclic voltammetry in the presence of reduced cytochrome c demonstrated that CcO was incorporated into the gel-supported ptBLM in a functionally active form.
Introduction
The lipid bilayer membrane plays a prominent role in many biologically relevant processes. Examples are signal transduction, transport of ions and molecules, biosynthesis, cell adhesion and recognition. Consequently, there is a great interest in model systems that would allow for a systematic study of such processes, preferably on a planar substrate. The idea of using macromolecules as a “cushion” to mimic the cytosol/cytoskeleton of the cell to create a hydrophilic space between the lipid membrane and solid support was first introduced by Ringsdorf and Sackmann.1,2 Since then polymer-supported lipid membranes have been widely used, as documented in several major review articles.3,4 Physical properties of the fluid lipid bilayer were investigated as well as processes taking place on the surface of the membrane. However, relatively few investigations of electrical properties of the lipid bilayer can be found in the literature. The reason is the difficulty of meeting the requirement known from patch clamp techniques, the giga-seal. Highly insulating polymer-supported membranes have been prepared, for example, when a very smooth surface of the hydrophilic polymer was achieved.5 Alternatively, specifically designed lipopolymers were employed, which were pre-oriented by the Langmuir–Blodgett technique.6–9 However, the polymer-supported lipid membrane having good electrical sealing properties remains a challenge.
A new approach in this direction is the use of a polymer layer as a substrate for the so-called protein-supported bilayer lipid membrane (ptBLM) developed previously on the basis of short linker molecules.10 This approach employs the protein immobilized on the substrate as a scaffold for the membrane reconstituted around the protein (Fig. 1). Consequently, this methodology begins with immobilizing the protein specifically onto the top layer of the polymer. This can be achieved by using a hydrogel as the polymer layer with mesh sizes smaller than the size of the protein. The gel is provided with a binding motif such as a nitrilotriacetic acid (NTA) functionality chelated with Ni+ ions designed to bind the proteinvia the his-tag technology.11,12 Since the protein is too big to penetrate the gel, it stays on the surface. If a lipid bilayer is then assembled around the protein, the formation of an electrically sealing protein–lipid layer could be expected. Electron and ion transport processes through the cytochrome c oxidase (CcO) have been observed in previous studies.13–15 In the present study a hydrogel consisting of poly(N-(2-hydroxyethyl)acrylamide-co-5-acrylamido-1-carboxypentyl-iminodiacetate-co-4-benzoylphenyl methacrylate) (P(HEAAm-co-NTAAAm-co-MABP)) was prepared by polymer analogous reactions that offers a NTA moiety to bind the CcO (Fig. 1). Optically transparent indium–tin oxide (ITO) layers were used as conducting substrates, different from previous studies using metal films.13–17 The advantage of ITO is that it allows the application of a number of surface-analytical techniques such as surface plasmon resonance and optical waveguide spectroscopy (SPR/OWS), and fluorescence techniques not applicable on metal films.
 |
| | Fig. 1 Schematics of the protein-supported bilayer lipid membrane (ptBLM) bound to the top layer of a hydrogel. Formation of the ptBLM started with immobilization of cytochrome c oxidase with the his-tag attached to SU I via the Ni complex to the NTA modified hydrogel P(HEAAm-co-NTAAAm-co-MABP). | |
Results and discussion
Preparation of the ITO layers
The ITO layers were prepared by DC sputtering on glass slides. The aim was to achieve good optical and electrical properties, as well as a low surface roughness of the layer. Sputtering conditions were varied regarding process pressure and argon/oxygen ratios. Sheet resistance, Rs, as a function of sputtering conditions is shown in Table 1. For the first three samples in Table 1 (1–3) the target was pre-sputtered using pure argon. The process pressure was decreased from Pb = 0.6 Pa to 0.3 Pa and the relative oxygen content was increased from 0 to 10 vol% Rs thereby decreased from 2k Ω sq−1 to 35 Ω sq−1. For samples 4–6 in Table 1 the target was pre-oxidized before deposition using a Pb of 1 Pa and 1 vol% oxygen. In the case of samples 4 and 5 Rs did not change much from 250 Ω sq−1. For the last sample (no. 6) oxygen was omitted leading to the best sheet resistance of 15 Ω sq−1.
Table 1 ITO sheet resistance, Rs, as a function of sputtering conditions, varying chamber pressure Pb and oxygen content
| Sample |
Pre-sputtering |
O2/vol% |
P
b/Pa |
R
s/Ω sq−1 |
| 1 |
1 Pa/pure Ar |
0 |
0.6 |
2k–2.5k |
| 2 |
5 |
0.6 |
250–500k |
| 3 |
10 |
0.3 |
35–85 |
| 4 |
1 Pa/Ar + 10 vol% O2 |
2 |
1 |
250–700 |
| 5 |
1 |
1 |
250 |
| 6 |
0 |
1 |
15–25 |
From this data it can be clearly seen that the electrical properties of the ITO layer depend on the state of the target before deposition. The target oxidation state changes with sputtering time and oxygen content. To describe this effect, the terms of ‘non-oxidized’ and ‘oxidized’ state of the target24 were introduced. In the first set of samples 1–3 in Table 1, only argon was used for pre-sputtering. Thereby the target was brought into the non-oxidized state leading to a brownish color of the ITO layer and a high sheet resistance of 2 to 2.5 k Ω sq−1. Too low oxygen content is known to reduce the transparency and conductivity of DC and RF sputtered ITO,25,26 whereas a high oxygen content still gives good transparency but a decreased conductivity. Therefore, in samples 2 and 3 oxygen was added with increasing partial pressure. This led to a decreasing resistance where the best result in terms of Rs was realized for sample 3 using 10 vol% of oxygen. Though sputtering had been started from a non-oxidized target, adding oxygen to the process seems to re-oxidize the target. In samples 4–6 re-oxidization of the target was achieved by pre-sputtering at a process pressure Pb of 1 Pa and 10 vol% oxygen. The sheet resistance became much lower compared to sputtering with a non-oxidized target. Starting deposition from the oxidized state of the target, (sample 6), resulted in the best values with Rs of 13–35 Ω sq−1. It can be concluded that after installation of the ITO target and before deposition, pre-oxidization of the target using an Ar–O2 mix is necessary. This way good electrical and optical properties of the layer were achieved. Consequently, samples were prepared on a routine basis from an oxidized target, pre-sputtered for 15 min using 10 vol% oxygen at 1 Pa process pressure. Samples were then sputtered in an atmosphere of pure argon at a process pressure of 1 Pa. Such samples had a reproducible Rs value of around 20 Ω sq−1.
Electrical and surface properties of ITO layers
All of the layers scanned by AFM show a surface pattern characterized by small spikes having a diameter of 15–20 nm and an average height of 7 nm. In general, the number of spikes decreased if an oxidized target was used for sputtering compared to the samples obtained from a non-oxidized target. Examples of AFM images of samples 3 and 6 from Table 1 are shown in Fig. 2. The number of spikes per area is significantly reduced in sample 6, but their height slightly increased. The formation of these spikes on the ITO surface could never be entirely avoided. So, ITO layers used on a routine basis always featured a surface morphology similar to Fig. 2B. ITO samples were also tested with respect to their electrochemical properties in PBS solution. Electrochemical impedance spectroscopy (EIS) was performed on bare ITO layers in PBS. EIS spectra at time intervals of 24 h and 48 h were recorded. An example of the evolution of these spectra is presented in Fig. 3. The dry ITO layers had a specific sheet resistance Rs of 40 Ω sq−1. The slides were pre-cleaned using isopropanol and UV–ozone treatment. The resistance obtained by EIS was relatively high compared to the sheet resistance, but decreased significantly within the first 24 h, whereas the capacitance increased to a value of 10–13 µF cm−2 (fitting results are summarized in Table 1). This value can most likely be attributed to the space charge capacitance Csc of the ITO semiconductor.27 The capacitance of the Gouy–Chapman layer is expected to be around 30 µF cm−2. Capacitance and resistance of the ITO layer in PBS did not change further for time intervals longer than 24 h, as seen from the data measured after 48 h. As an explanation a highly resistive surface layer is considered at the beginning (note that the sheet resistance reflects the bulk phase of ITO whereas EIS measures the interface toward PBS). As the ITO layer is submersed in PBS a hydration process sets in during which water is taken up by the ITO layer, reaching an equilibrium state after a certain time. With a dielectric constant of 80 for water, the capacitance is expected to increase in agreement with our observation. The uptake of water molecules can be explained in terms of the porous structure of the bulk ITO, formed during the sputtering process. Water diffuses through the pores into the ITO layer, which is equivalent to observations made for aluminium oxide layers sputtered on glass substrates.28 This assumption is also supported by the AFM images of the DC sputtered ITO surface (Fig. 2).
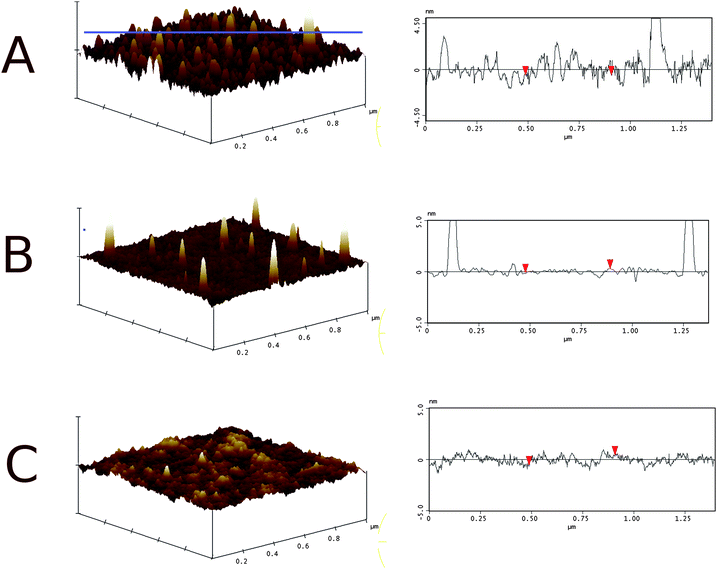 |
| | Fig. 2 3D surface profile (left) and corresponding line-scan (right) of DC sputtered ITO: (A) sample 3 and (B) sample 6 from Table 1. The hydrogel coated sample (C) is also shown. The electrode surface is smoothened and the spikes are covered by the gel. The z-scale on the left is 10 nm. The location of the line scan on the left is indicated by the horizontal line in (A) and was oriented in the same manner in all three samples. | |
 |
| | Fig. 3 (A) Bode plot and (B) frequency normalized admittance of the impedance spectra of (●) bare ITO electrodes in PBS measured after (□) 24 h and (▲) 48 h and their corresponding fit curves (circuit I in Table 2). The sheet resistance of the ITO layer was 40 Ω sq−1. | |
Hydrogel-supported ptBLMs on ITO layer measured by EIS
The ITO surface was coated with a layer of the copolymer as described in the Experimental part. The hydrogel was obtained as a highly crosslinked layer from P(PFPA-co-MABP) in three reaction steps as depicted in Fig. 4. The precursor copolymer P(PFPA-co-MABP) was prepared by free radical polymerization from 98 mol% PFPA and 2 mol% MABP, for details see the Experimental part. In the first step, this precursor copolymer was spin-coated on a benzoylphenyl (BP)-silane functionalized ITO surface and UV-crosslinked. The thickness of the dry copolymer layer was 60 nm measured by AFM and ellipsometry. Thereby, the surface of the ITO layer was flattened and a chemically durable surface was obtained with high density of the hydrophobic pentafluorophenol active ester. These functional groups exhibit a high reactivity towards amines and prevent the swelling by water during the coupling of amino-NTA in aqueous media in the second step. This resulted in a NTA-functionalization predominantly at the polymer–water interface. The subsequent reaction with ethanolamine transformed the hydrophobic copolymer into the hydrophilic hydrogel. Such a coating fulfills the criteria mentioned in the introduction, namely providing a stable, smooth, hydrophilic, and reactive surface. Stability and surface roughness are determined by molecular weight of the polymer and the extend of crosslinking (e.g. the amount of crosslinker groups and the crosslinking irradiation dose). The employed copolymer was optimized resulting in a durable coating of low roughness. AFM measurements showed the spikes of the ITO layer to be completely covered by the polymer layer (Fig. 2C). The coating had a low mean roughness (RMS = 0.4 nm) and a peak-to-valley height of 4 nm, a highly important prerequisite for the assembly of a dense lipid membrane. EIS spectra showed a considerable increase in the capacitance and a decrease in the resistance. This can be explained by the presence of the hydrophobic pentafluorophenol active ester functionalities. Before the copolymer was converted into a hydrogel, the low capacitance is dominated by the thin dielectric layer. When the active ester was functionalized with hydrophilic groups such as NTA and OH, thus converting the copolymer into a hydrogel, water molecules could enter the hydrogel together with ions from the buffer solution to form a Gouy–Chapman electrical double layer. Since water has a high dielectric constant the capacitance of the hydrogel layer increases drastically. Hence, the capacitance of ∼15 µF cm−2 is again dominated by Csc, as described above. The NTA functionalities were converted to the Ni–NTA chelate by immersion of the gel layer into a buffered NiSO4 solution. The excess Ni was removed by rinsing with an acetate buffer solution (pH = 5.5). After that a solution in dodecyl β-D-maltoside detergent (DDM) of CcO from Paracoccus denitrificans with a his-tag attached to the C-terminus of subunit I was added to the bathing solution. As seen in Fig. 5 the impedance spectrum did not change much, although the binding of CcO could be detected by waveguide measurements described further below. However, when the phospholipid (DiPhyPC) solubilized in DDM was added together with biobeads, the resistance increased by two orders of magnitude to reach values 1–5 MΩ cm2. Fig. 5 shows a typical example. This indicates the insertion of lipid bilayer patches between the CcO molecules, or in other words the formation of a ptBLM. EIS data are collected in Table 2. The resistance of 1 to 5 MΩ cm2 is in accordance with results obtained for ptBLMs on smooth gold films without a gel layer.29 The resistance is known to be a very critical parameter to indicate a defect-free lipid bilayer. Values in the order of magnitude of MΩ cm2 are known from freely suspended bilayer lipid membranes (BLMs) and tethered bilayer lipid membranes (tBLMs),30,31 whereas for polymer-supported bilayers such high sealing resistances were hard to achieve. The capacitance does not change much because it is dominated by the capacitance of the space charge region as well as the protein layer, which are expected to be in the same order of magnitude. The high surface density of proteins causes a higher capacitance of the protein/lipid layer as compared to a lipid bilayer without proteins. The high resistance after dialysis, however, indicates that CcO molecules were bound mainly to the surface layer rather than in the meshes of the gel layer. Only protein molecules attached to the surface can form a closed protein–lipid layer as requested for a ptBLM.
 |
| | Fig. 4 Synthesis of the precursor polymer P(PFPA-co-MABP) and its conversion into the PHEAAm-hydrogel with attached NTA groups (P(HEAAm-co-NTAAAm-co-MABP)). | |
 |
| | Fig. 5 Frequency response presented as (A) Bode and (B) frequency normalized admittance plot of the ITO coated with the P(PFPA-co-MABP) precursor; (●) before and (□) after conversion to P(HEAAm-co-NTAAAm-co-MABP) using amino-NTA and ethanolamine. Also shown are the fits (solid lines) using equivalent circuit I in Table 2. | |
 |
| | Fig. 6 Bode plots (A) and frequency normalized admittance (B) of the Ni2+–NTA modified P(HEAAm-co-NTAAAm-co-MABP) on ITO before (●) and after (□) binding of CcO; (▲) after in situdialysis to form the lipid bilayer around the protein. Solid lines show the fitted curves using circuit I (Table 2). For the fit in (▲) equivalent circuit II in Table 2 was used. | |
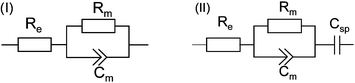
Table 2 Resistance and capacitance values derived from nonlinear least-squares fit to the impedance spectra of different preparation steps using the equivalent circuits (I) and (II)
| ITO samples at different preparation steps |
R
m/kΩ cm2 |
C
sp/µF cm−2 |
|
Fitted using equivalent circuit (II), otherwise circuit (I) was employed.
|
| Bare ITO in PBS |
800–1190 |
9 |
| After 24 h |
15–56 |
12–15 |
| After 48 h |
13–64 |
12–15 |
|
P(PFPA-co-MABP) coated |
>6 × 104 |
0.04 |
| Conversion of P(PFPA-co-MABP) to hydrogel |
15–39 |
13–16 |
|
CcO binding |
70–110 |
14–16 |
| ptBLM formationa |
(0.8–5) × 103 |
12–15 |
| Addition of 50 µM cyt cred after ptBLM formation |
(0.1–0.7) × 103 |
15–19 |
| removal of cyt c using PBS |
(0.5–1) × 103 |
13–15 |
Oriented immobilization of the CcO and reconstitution into a ptBLM investigated by SPR/OWS
EIS is not designed to follow the binding of the CcO to the NTA functionalized polymer layer. The formation of the lipid bilayer can be deduced from these measurements, but only indirectly. More information can be expected from SPR/OWS measurements, which were performed on a thick (680 nm) ITO layer as the wave guiding media. SPR/OWS is a multi-mode technique with TM and TE modes, i.e. angle scans obtained from p-polarized and s-polarized light, respectively. Binding as well as lipid reconstitution could be clearly seen as a shift of the TM3 (p-polarized) and the TE2 mode (s-polarized) in the angle spectra in Fig. 7. The effective refractive indices Neff of the propagating TM3 and TE2 (Table 3) are calculated from the angle spectra using eqn (1)| | 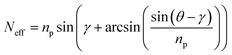 | (1) |
where Θ is the measured angle and γ the prism angle. np denotes the refractive index of the prism (see Experimental).
 |
| | Fig. 7
SPR/OWS angle spectra of (A) p-polarized and (B) s-polarized light (632 nm) of (solid line) Ni2+–NTA modified P(HEAAm-co-NTAAAm-co-MABP) on ITO before and (dashed line) after binding of CcO; (dotted line) after in situdialysis to form the lipid bilayer around the protein. | |
Table 3 The change of the effective refractive index Neff of the TE2 and TM3 modes after each preparation step calculated using eqn (1)
| |
dNeffTM3 |
dNeffTE2 |
|
Ni2+–NTA modified P(HEAm-co-TAAm-co-MABP) |
0 |
0 |
| After CcO adsorption |
2.971 × 10−3 |
6.432 × 10−3 |
| After in situdialysis for the formation of the BLM |
5.875 × 10−3 |
8.243 × 10−3 |
Changes of dNeff from one layer to the next give information about the anisotropy of the layers. From the angle scans it is already obvious that the two modes shift differently for each layer formation. In order to obtain a more quantitative information dNeff values of the two modes were simulated for the isotropic case as a function of the thickness of the ITO layer. The thickness of the gel-support was set to 60 nm (see Experimental). From this so-called sensitivity curve (Fig. 8) the expected absolute shift dNeff of both modes, TM3 and TE2 at a particular thickness d of the ITO layer, can be derived. In the experiments performed here (d = 680 nm) the two modes should shift at a ratio TM3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TE2 of 2.4:1 provided the layers are isotropic.
:TE2 of 2.4:1 provided the layers are isotropic.
 |
| | Fig. 8 (A) Simulated shifts dNeff of the effective refractive index caused by an isotropic adlayer (n = 1.47, d = 11 nm) on ITO calculated for the TE and TM modes as a function of the thickness of the ITO layer (n = 1.9065): (■) TM1, (▲) TM2, (◆) TM3, (△) TE0, (○) TE1, and (□) TE2. The arrow indicates the thickness of the ITO layer used on a routine basis. (B) dNeff for the TM and TE optical waveguide modes after each preparation step of the ptBLM on P(HEAAm-co-NTAAAm-co-MABP). The experimental dNeff values were derived from the Neff in Table 3. | |
The actual dNeff values derived from the Neff in Table 3 show different behavior as expected for an anisotropic system, Fig. 8 presents actual and simulated dNeff of the protein and the protein/lipid layer. In both cases the two modes, TE2 and TM3, shift at a ratio different from the theoretical one. The TE2 mode shifts even stronger than the TM3 mode in contrast to what is expected from simulation data. This result can only be explained by an anisotropy of both the CcO and the lipid/CcO layer. However, with the dipole moment directed along the x-axis of the CcO a shift of the TE mode indicates the CcO molecules being arranged more parallel to the surface rather than perpendicular. They seem to rearrange into a more upright position as deduced from the difference between TE and TM modes decreasing after insertion of the lipids as required for a well-ordered planar lipid bilayer.32
Activity of the CcO in the gel-supported ptBLM in the presence of cyt c investigated by cyclic voltammetry
CVs of the hydrogel were first measured in the presence of reduced cyt c before adsorption of CcO and formation of the ptBLM. The CVs recorded immediately after the addition of cyt c showed oxidation/reduction peaks with a peak separation of 160 mV, where the peak height did not change with time (Fig. 9(A), solid line). From this we can deduce that cyt c was able to penetrate the hydrogel layer without any delay time so that it could be oxidized and reduced at the electrode.
 |
| | Fig. 9
Cyclic voltammogram (A) of 50 µM reduced cyt c at different steps of the sample preparation; P(HEAAm-co-NTAAAm-co-MABP) (solid line) before immobilization of CcO, (dashed line) after immobilization and re-constitution of CcO into the lipid bilayer, (dotted line) after rinsing with fresh buffer. The scan rate was 50 mV s−1. (B) Impedance spectra of CcO (▲) after re-constitution into the lipid bilayer, (○) activation using 50 µM reduced cyt c and (□) subsequent flushing using PBS. Solid lines represent fits using equivalent circuit I in Table 2. | |
In the next experiment reduced cyt c was added at the same concentration to the NTA- and aminoethanol-functionalized hydrogel layer after adsorption of CcO and the formation of the ptBLM. In this case the CV showed an increase of the cathodic current as well as an increase in the peak separation to 364 mV between cathodic and anodic peaks (Fig. 9(A), dashed line). The amplification of the cathodic current is an indication of the catalytic activity of the CcO. The reduced cyt c added to the bulk phase is oxidized by the enzyme and hence has to be reduced repeatedly at the electrode. The result is an increased, so-called catalytic current. The higher peak separation, on the other hand, indicates the barrier properties of the lipid layer. In this case, the cyt c had to penetrate not only the gel but also the protein–lipid layer. The diffusion through both layers is reversible as shown in Fig. 9(A) (dashed line). Only the charging current was left after rinsing with fresh buffer solution. In summary, this experiment demonstrated that the CcO immobilized within the hydrogel-supported ptBLM was catalytically active.
Activity of the CcO in the gel-supported ptBLM in the presence of cytochrome c (cyt c) investigated by EIS
Finally EIS was conducted in order to confirm the results described in the previous paragraph (Fig. 9(B)). We began with a gel-supported ptBLM after reconstitution of the lipid membrane showing a resistance of ∼5 MΩ indicating good sealing properties. After addition of reduced cyt c the resistance decreased to 0.7 MΩ. The resistance of the ptBLM could be restored by flushing the cyt c away with fresh buffer (Table 2). The sharp drop of the resistance as a function of cyt c was explained before in the case of the ptBLM on smooth Au surfaces not only in terms of the electron exchange current of cyt c,16 but also the proton current across the lipid bilayer taking place as a consequence of the redox reactions contributes to the decrease of the resistance. Hence the overall effect is more pronounced than in the case of cyclic voltammetry. In any case, the data are very well comparable to previous measurements with the ptBLM on smooth Au surfaces.
In order to underscore the assertion mentioned above that the protein–lipid mixture forms a closed layer on top of the hydrogel, we used the his-tagged light harvesting protein LHCIIb from pea as a fluorescent protein to form the ptBLM under otherwise identical conditions (Fig. 10).
 |
| | Fig. 10 (A) Example of the fluorescence spectrum of the trimeric LHCIIb in a gel-supported ptBLM system on ITO. (B) Laser scanning microscopy (LSM) image. The fluorescence intensity in the bleached area in (B) did not recover with time. (C) LSM image of the LHCIIb/CcO mixed layer (0.1 mol% LHCIIb). | |
The laser scanning microscopy (LSM) image showed a uniform fluorescence indicating a closed layer rather than aggregates or clusters. Fluorescence recovery after photobleaching (FRAP) was not obtained as expected for a protein immobilized on the surface. The fluorescence spectrum showed the characteristic features of LHCIIb as described in ref. 33 (Fig. 10). Dilution of the LHCII solution with his-tagged CcO to a concentration of 0.1 mol% LHCIIb resulted in an LSM image with single fluorescent spots in an otherwise non-fluorescent layer (Fig. 10(B)), also indicating a uniform distribution of the proteins bound to the surface rather than penetrating the hydrogel.
Conclusion
We conclude from these results that the his-tagged proteins are indeed immobilized on top of the hydrogel layer. Otherwise the dramatic increase of the resistance after in situdialysis by two orders of magnitude would not be possible (see Table 2). Proteins immobilized in the cavities formed by the hydrogel would never reconstitute into a closed lipid bilayer. Moreover, the reversible decrease of this resistance by adding the substrate cyt c in the reduced form and flushing it away is very characteristic for the flow of charged particles such as electrons and protons through a bilayer lipid membrane (Fig. 10 and Table 2). Smaller proteins such as cyt c, on the other hand, appear to be able to penetrate so as to reach the electrode as seen from the CVs. From the relative size of the two proteins, 4 and 9 nm for cyt c and CcO, respectively, an estimate of the mesh size of the hydrogel can be made to be approximately 5–7 nm. Another argument in favour of the top layer of protein–lipid molecules was derived from SPR/OWS measurements. The use of ITO as a conducting and wave guiding substrate allowed us to address the question of the anisotropy of the surface layers. A well-ordered lipid bilayer should exhibit different optical properties in different spatial directions. The same argument holds to a lesser extent for an ordered and oriented protein monolayer. The experimental proof of this anisotropy is hard to achieve and hence information about the proper arrangement of a protein/lipid layer is scarce. Owing to their multi-mode approach waveguide measurements have the potential to access these properties. The advantage of ITO layers is that they can be used as a waveguide as well as a substrate for electrochemical measurements. However, preparation conditions need to be optimized carefully. The thickness needs to be compromised vs. stability and surface roughness of the ITO. Surface roughness that was deteriorated by spike structures could successfully be overcome by coverage with the hydrogel. The polymer layer in turn had to be kept at a thickness of around 60 nm in order to ensure a smooth surface for the protein/lipid layer. About 60 nm layer thickness is sufficient to mimic the submembrane space. Small molecules such as cyt c penetrate easily. If, on the other hand, the large his-tagged proteins would be inside the cavities of the hydrogel, they would be immobilized in a random orientation such that different dNeff values could never be measured by SPR/OWS indicating the anisotropy, particularly after insertion of the lipid bilayer patches. Finally, fluorescence measurements are also consistent with the top layer of protein–lipid molecules. FSM images show an evenly distributed intensity just as fluorescent proteins in an intact bilayer lipid membrane. The fluorescence intensity of LHCII in the ptBLM did not recover after bleaching, which is consistent with the notion of proteins immobilized in-between the bilayer patches. Hence the concept of the ptBLM proved to be useful in the context of polymer-supported planar lipid bilayers. The hydrogel-supported ptBLMs based on a polymer with mesh sizes smaller than the size of the protein were shown to result in very robust polymer-supported planar lipid bilayers, exhibiting good electrical sealing properties. Cytochrome c oxidase could be shown to be incorporated in a functionally active form.
Experimental
4-(3-Triethoxysilyl)propyloxybenzophenone
18 (BP-silane), pentafluorophenyl acrylate19 (PFPA) and 4-benzoylphenyl methacrylate20 (MABP) were prepared according to the literature. Azodiisobutyronitrile (AIBN) (Sigma Aldrich, Germany) was recrystallized from methanol. Dioxane (Sigma Aldrich, Germany) was distilled over calcium hydride.
The reactive copolymer poly(pentafluorophenol acrylate-co-4-benzoylphenyl methacrylate) (P(PFPA-co-MABP)) was polymerized from 98 mol% PFPA and 2 mol% MABP by free radical polymerization with 0.2 mol% AIBN in dioxane at 60 °C for 48 h. It was precipitated in methanol and dried in high vacuum.
The reactive copolymer was obtained with a yield of 78% after precipitating it three times from benzene in methanol. The molecular weight Mn was determined by GPC (tetrahydrofuran as solvent and poly(methylmethacrylate) as standard) to be ∼14.000 g mol−1 and the molecular weight distribution was ∼2.1. The 1H-NMR shows the peaks of the PFPA and the MABP with a ratio of 98:2 as intended by the monomer mixture. 1H-NMR (700 MHz, CD2Cl2): δ [ppm] = 7.45 (9H, br m, benzophenone), 3.11 (1H, br s, CH, backbone), 2.15 (2H, br s, CH2, backbone), 1.48 (3H, br m, CH3, backbone).
Preparation of ITO layers on glass slides
The ITO layer was deposited on float glass (Menzel, Braunschweig, Germany). Glass slides were cleaned in piranha solution (H2O:H2SO4:H2O2, 5:1:1, v/v) and rinsed using DI water. ITO was deposited by DC magnetron sputtering using a Balzers sputtering system (Oerlikon Balzers Lichtenstein). The target used was a 3 inch In2O3:SnO2 (90:10) (MaTeck, Juelich, Germany). Series of slides were always prepared on the same day. After deposition under each parameter set used the sheet resistance Rs was measured by a four-point probe method according to Van-der-Paw.22 For the samples used on a routine basis, the process pressure was Pb = 5.5 × 10−6 mbar. The ITO target was pre-treated without the samples under following conditions: 15 min sputtering time using an argon/oxygen mix (10 vol% O2) at a gas pressure Pb of 1.1 Pa and a DC power of 100 W. Afterwards 100 nm ITO was sputtered on top of the glass slides using a pure Ar atmosphere without any oxygen at 0.27 Pa and 100 W. The deposition time was usually 10–15 min and the layer thickness, measured by step-profiler and ellipsometry, was 80–120 nm. The morphology of the ITO surface was analyzed by AFM.
Preparation of the gel layer
The ITO coated glass slides were functionalized with the BP-silane as described earlier.18 On top of the silane layer the reactive copolymer P(PFPA-co-MAPB) was spin-coated from chloroform (1 wt%), dried overnight at 50 °C in a vacuum and cross-linked by photopolymerization with a wavelength of 254 nm and a total energy of 60 J cm−2.
Functionalization with NTA
The polymer-coated ITO samples were incubated for 2 h in an aqueous 0.15 M nitrilotriacetic acid (amino-NTA) solution (pH 9.8, 0.5 M K2CO3 buffer) and after 2 h ethanolamine (80 mM) was added to the same solution. The samples were left for a further 30 min in the solution and subsequently rinsed using Milli-Q water.
Binding of CcO and reconstitution of a lipid bilayer membrane
CcO from P. denitrificans with a his-tag at the C-terminus of subunit I was prepared according to the literature.21
The ITO slides were immersed for 30 min in 40 mM NiSO4 in acetate buffer (50 mM, pH 5.5). The excess Ni was removed by brief rinsing using the same acetate buffer without Ni. Then CcO dissolved in dodecyl β-D-maltoside (DDM)-phosphate-buffer (K2HPO4 0.1 M, KCl 0.05 M, pH = 8, 0.1% DDM) was adsorbed to the NTA-functionalized surface at a final concentration of 100 nM. Bio-Beads were added to the lipid detergent containing phosphate buffer (K2HPO4 0.1 M, KCl 0.05 M, pH 8, diphytanoylphosphatidylcholine (DiPhyPC) 0.05 mg ml−1, 0.1% DDM) to remove the detergent and to form a lipid bilayer.
Ellipsometry measurements were performed using an EP3 System (Nanofilm Technologies, Göttingen, Germany). Refractive index and absorption coefficient of the ITO layer were measured by multiple-angle scan in the region of the Brewster angle, which was found to be 62° for the ITO layers used in this work. The data were analyzed using the EP3 View software (v. 2.3). The refractive index ng of the glass substrate was taken to be 1.55. The thickness of the ITO layer was also measured by profilometer. Therefore, samples have been partially masked before deposition using tape. The partially coated glass slides were scanned across the step from the coated to the uncoated region. The average step height was taken as the thickness of the ITO layer.
Electrochemical measurements were taken in a three-electrode configuration where the ITO slide was used as the working, a home-made Ag/AgCl (sat. KCl) as the reference, and a platinum wire as the counter electrode. Electrochemical impedance spectra and cyclic voltammetry measurements were performed using Autolab instrument PGSTAT302 (Eco Chemie, Utrecht, The Netherlands) equipped with an FRA2 module for frequency response analysis, an ECD-module amplifier for low currents and a SCAN-GEN module for analogue potential scanning. Spectra were recorded in the frequency range from 100 kHz to 3 mHz using an amplitude of 10 mV. Resulting spectra were analyzed by Zview (Version 2.6, Scribner Associates, Southern Pines, NC) by complex nonlinear fitting of the data to a model circuit. The scan rate of the CV measurements was fixed to 50 mV s−1.
SPR/OWS was performed in a custom made setup described previously using the Kretschmann-configuration.23 The glass slide (LaSFN9 glass from Hellma Optic, Jena, refractive index n = 1.8385 at 633 nm) was optically matched to the base of a 45° glass prism (LaSFN9). Monochromatic light from a He/Ne laser (Uniphase, San Jose, CA, λ = 632.8 nm) was directed through the prism and collected by a custom made photodiode detector. The glass slide was provided with a multilayer system starting with a 41 nm thick gold layer electrothermally evaporated on top of 2 nm thick Cr adhesion layer. A 680 nm thick ITO layer was then sputtered onto the gold layer under the conditions described above and the surface was spin-coated with P(HEAAm-co-NTAAAm-co-MABP). ITO served as the guiding media as well as the working electrode. The Neff values of the gel-supported tBLM are calculated and collected in Table 3. Change of Neff (dNeff) for varying ITO thickness was simulated using n = 1.9065 for ITO taken from ellipsometry measurements. The simulation software package used was CAMFR (CAvity Modelling Framework, INTEC, Universiteit Gent).
Atomic force microscopy was performed using the AFM Dimension 3100CL Olympus (Veeco Instruments Inc., NY) in tapping mode with a silicon cantilever. To verify that the selected location was representative for the whole ITO surface, randomly chosen spots (1 × 1 µm) out of a 1 cm2 area were scanned and compared. Only scans around the center of the sample surface were comparable to each other. For that reason the measurement was always performed at the center of the sample surface choosing the highest scanning resolution (512 × 512 lines). The height profile images were analyzed in terms of root mean square (RMS) and peak-valley height (Nanoscope software v2.5r, Veeco).
Preparation of reduced cyt c
The stock solution of reduced bovine heart cyt c (Sigma Aldrich, St Louis, MO) was prepared by adding 5 mg of sodium dithionite to an aqueous solution of 40 mg cyt c in 1 ml PBS. The reducing agent was removed subsequently by gel filtration utilizing a Sephadex column (G-25 M, GE Healthcare Bio-Science AB, Uppsala, Sweden).
Trimeric light harvesting complex LHCIIb from pea (Pisum sativum) with the his-tag attached to the C-terminus was expressed and purified according to ref. 34. Laser scanning microscopy (LSM) images were recorded on a commercial setup (Carl Zeiss, Jena, Germany) consisting of the modules LSM510, ConfoCor 2 and an inverted microscope model Axiovert 200. The LHCIIb complex was excited using an argon laser (488 nm). Emission light was collected using a 40× water immersion objective with numerical aperture of 1.2 and was detected after an LP650 long pass emission filter. A spectrometer (SpectraPro 2300i, Acton Research, Acton, MA) with a charge-coupled device (CCD) camera was attached to the confocal microscope to record the fluorescence spectra (Fig. 10) of the reconstituted LHCIIb in the gel-supported ptBLM system on ITO.
Acknowledgements
Partial support for this work was provided by ZIT, Center of Innovation and Technology of Vienna.
Notes and references
- M. Kuhner, R. Tampe and E. Sackmann, Biophys. J., 1994, 67, 217–226 CAS.
- J. Simon, M. Kuhner, H. Ringsdorf and E. Sackmann, Chem. Phys. Lipids, 1995, 76, 241–258 CrossRef CAS.
- E. Sackmann, Science, 1996, 271, 43–48 CrossRef CAS.
- M. Tanaka and E. Sackmann, Nature, 2005, 437, 656–663 CrossRef CAS.
- H. Hillebrandt, G. Wiegand, M. Tanaka and E. Sackmann, Langmuir, 1999, 15, 8451–8459 CrossRef CAS.
- S. Schiller, A. Reisinger-Friebis, H. Götz, C. J. Hawker, C. W. Frank, R. Naumann and W. Knoll, Angew. Chem., Int. Ed., 2009, 48, 6896–6899 CrossRef CAS.
- V. Nikolov, J. Lin, M. Merzlyakov, K. Hristova and P. C. Searson, Langmuir, 2007, 23, 13040–1304 CrossRef CAS.
- C. A. Naumann, O. Prucker, T. Lehmann, J. Ruhe, W. Knoll and C. W. Frank, Biomacromolecules, 2002, 3, 27–35 CrossRef CAS.
- O. Purrucker, A. Fortig, R. Jordan and M. Tanaka, ChemPhysChem, 2004, 5, 327–335 CrossRef CAS.
- F. Giess, M. G. Friedrich, J. Heberle, R. Naumann and W. Knoll, Biophys. J., 2004, 87, 3213–3220 CrossRef CAS.
- J. Arnau, C. Lauritzen, G. E. Petersen and J. Pedersen, Protein Expression Purif., 2006, 48, 1–13 CrossRef CAS.
- E. Hochuli, H. Dobeli and A. Schacher, J. Chromatogr., A, 1987, 411, 177–184 CrossRef CAS.
- M. G. Friedrich, J. W. F. Robertson, D. Walz, W. Knoll and R. L. C. Naumann, Biophys. J., 2008, 94(9), 3698–3705 CrossRef CAS.
- C. Nowak, M. G. Santonicola, D. Schach, J. Zhu, R. B. Gennis, S. Ferguson-Miller, D. Baurecht, D. Walz, W. Knoll and R. L. C. Naumann, Soft Matter, 2010 10.1039/c0sm00160k.
- K. Ataka, F. Giess, W. Knoll, R. Naumann, S. Haber-Pohlmeier, B. Richter and J. Heberle, J. Am. Chem. Soc., 2004, 126, 16199–16206 CrossRef CAS.
- M. G. Friedrich, M. A. Plum, M. G. Santonicola, V. U. Kirste, W. Knoll, B. Ludwig and R. L. C. Naumann, Biophys. J., 2008, 95, 1500–1510 CrossRef CAS.
- M. G. Friedrich, F. Giess, R. Naumann, W. Knoll, K. Ataka, J. Heberle, J. Hrabakova, D. H. Murgida and P. Hildebrandt, Chem. Commun., 2004, 2376–2377 RSC.
- M. Gianneli, R. F. Roskamp, U. Jonas, B. Loppinet, G. Fytas and W. Knoll, Soft Matter, 2008, 4, 1443–1447 RSC.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- R. Toomey, D. Freidank and J. Rühe, Macromolecules, 2004, 37, 882–887 CrossRef CAS.
- S. Lucioli, K. Hoffmeier, R. Carrozzo, A. Tessa, B. Ludwig and F. M. Santorelli, Neurogenetics, 2006, 7, 51–57 CrossRef.
- A. Ramadan, R. D. Gould and A. Ashour, Thin Solid Films, 1994, 239, 272–275 CrossRef CAS.
- W. Knoll, Annu. Rev. Phys. Chem., 1998, 49, 569 CrossRef CAS.
- F. Kurdesau, G. Khripunov, A. F. da Cunha, M. Kaelin and A. N. Tiwari, J. Non-Cryst. Solids, 2006, 352, 1466–1470 CrossRef CAS.
- Y. S. Jung, D. W. Lee and D. Y. Jeon, Appl. Surf. Sci., 2004, 221, 136–142 CrossRef CAS.
- H. Lin, J. Yu, S. Lou, J. Wang and Y. Jiang, Proc. Soc. Photo-Opt. Instrum. Eng., 2007, 672243-1–672243-5.
- S. Gritsch, P. Nollert, F. Jähnig and E. Sackmann, Langmuir, 1998, 12, 3118–3125 CrossRef.
- R. F. Roskamp, I. K. Vockenroth, N. Eisenmenger, J. Braunagel and I. Koper, ChemPhysChem, 2008, 9, 1920–1924 CrossRef CAS.
- F. Giess, M. G. Friedrich, J. Heberle, R. L. Naumann and W. Knoll, Biophys. J., 2004, 87, 3213–3220 CrossRef CAS.
- L. Becucci, M. R. Moncelli and R. Guidelli, Langmuir, 2003, 19, 3386–3392 CrossRef CAS.
- H. He, J. W. F. Robertson, J. Li, I. Karcher, S. M. Schiller, W. Knoll and R. Naumann, Langmuir, 2005, 21, 11666–11672 CrossRef CAS.
- Z. Salamon and G. Tollin, Biophys. J., 2001, 86, 2508–2516.
- J. Liu, R. Lauterbach, H. Paulsen and W. Knoll, Langmuir, 2008, 24, 9661–9667 CrossRef CAS.
- S. Hobe, I. Trostmann, S. Raunser and H. Paulsen, J. Biol. Chem., 2006, 281, 25156–25166 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.