Aqueous phase behavior of salt-free catanionic surfactants: the influence of solubility mismatch on spontaneous curvature and balance of forces
Bruno F. B.
Silva
a,
Eduardo F.
Marques
*a and
Ulf
Olsson
b
aCentro de Investigação em Química, Department of Chemistry and Biochemistry, Faculty of Science, University of Porto, Rua do Campo Alegre, n° 687, P 4169-007, Porto, Portugal. E-mail: efmarque@fc.up.pt
bPhysical Chemistry, Centre for Chemistry and Chemical Engineering, Lund University, P.O. Box 124, SE-221 00, Lund, Sweden
First published on 11th October 2010
Abstract
In this paper, we investigate the phase behavior and microstructure for a series of salt-free catanionic surfactants of the type Cm+Cn− with varied chain length mismatch (m ≠ n), using light microscopy, DSC, turbidity, surface tension, SAXS and SANS. The compounds consist of alkyltrimethylammonium alkylsulfonates, denoted by TAmSon. Depending on the asymmetry between both ions, three regimes can be identified: (i) weakly asymmetric; (ii) approximately symmetric; and (iii) highly asymmetric. For the TA16Son compounds, with n = 8 and 9 (weakly asymmetric), the surfactant forms a lamellar phase in water, however, with a striking miscibility gap. This miscibility gap is a consequence of the concentration dependent bilayer charge density. For n = 8, also a temperature-dependent vesicle-to-micelle transition at low surfactant concentration is observed. When the mismatch is low (n = 10) only a non-swelling lamellar phase is formed (approximately symmetric regime). For high mismatch, (n = 6 and 7) an extensive micellar phase is obtained – highly asymmetric regime. Conversely, for the TAmSo8 compounds, where m = 12 and 14, the unconventional lamellar miscibility gap and vesicle-micelle transition are again present. These findings are rationalized by considering the effect of film charge density—arising from the chain solubility difference—on the spontaneous curvature and balance of colloidal forces. The type of phase behavior reported here should be extensive to other families of salt-free catanionic amphiphiles, where an appropriate tuning of the solubility mismatch can allow the control of self-assembly.
1. Introduction
Swelling bilayer-forming surfactants are important materials for the building of mimetic models for biomembranes and in applications such as the formulation of microemulsions,1,2 multilayered films,3 nanoreactors4 and molecular transporters,5 to cite but a few. In addition, if these molecules show some type of stimuli-responsive self-assembly in aqueous or non-aqueous solvents,6 further potential is added to their practical relevance, e.g. in the rheological control of colloidal materials. In this context, salt-free catanionic surfactants are a versatile class of swelling compounds whose aggregation behavior is easily tunable by the manipulation of their chemistry and of physical parameters such as temperature and salt.Catanionic surfactants are complex salts of the type Cm+Cn− that result from the stoichiometric pairing of a cationic, Cm+X−, and an anionic, Cn−X+, amphiphile (where m and n indicate the chain length), with inorganic salt removal.7–11 In contrast to the more general catanionic mixtures, which also contain inorganic salt (X+X−) and excess ionic amphiphile, catanionic surfactants give rise in water to true binary systems, easier to characterize and to model thermodynamically than the mixtures (pseudo-ternary systems). Any surface charge on the surfactant film arises solely from differences in chain solubility and this will lead to long screening lengths and considerable impact on film properties11 and global self-assembly.7,12–17
Most of the catanionic amphiphiles studied so far have identical or similar chain lengths, |m − n| ≤ 4. If in addition the chains are long enough, m > 12 or n > 12, these surfactants display high Krafft points, a small solubility in water and a classic swelling behavior.8,18 However, if the chain lengths are made significantly different, typically |m − n| > 4, two factors may come into play. First, a higher configurational entropy caused by the chain length mismatch is introduced in the crystalline lattice, leading to a relative destabilization of the crystal19 and, hence, lower Krafft points. Secondly, if the smaller chain surfactant is more soluble than the longer one, a net surface charge on the catanionic film arises which, furthermore, is dependent on the total surfactant concentration.15,20 These effects may lead to important consequences on the aggregation behavior both at high and low surfactant volume fraction.
A few reports have been published on salt-free Cm+Cn−amphiphiles, showing that these compounds form multifaceted self-assemblies, such as normal and reverse micelles, vesicles, onion structures and various types of liquid crystals.8,15,16,21–24 Persson et al.,25 for instance, have studied the phase behavior of n-alkylpyridinium octylsulfonates in water, while Eastoe et al.26 investigated the micellization of alkyltrimethylammonium dodecylsulfates. Equimolar surfactant-hydrotrope complexes, where the small inorganic ion is replaced with an organic one in analogy with catanionic surfactants, have also been extensively investigated.27–37
However, no study has yet addressed in a systematic way the effect of the Cm+/Cn− solubility mismatch on the aggregation behavior over the full concentration range, and provided a generalized picture for the observed self-assembly. Recently, we have studied compounds with fixed asymmetry in chain length (m = 16 and n = 8) and different headgroup chemistry.38 In the current work, we keep the headgroup chemistry fixed and vary instead the chain solubility difference. For that purpose, we have prepared several alkyltrimethylammonium alkylsulfonates, comprising the compounds designated by TA16Son, where n = 6–10 (fixed m = 16), and by TAmSo8, where m = 12 and 14 (fixed n = 8).
For these surfactants, the higher solubility of the anionic part, Son− originates a weakly charged film, typically a bilayer, and an electrostatic force whose magnitude is concentration-dependent. Considering that attractive van der Waals and short-range repulsive forces also come into play, the net balance of forces may result in an unusual swelling behavior, with a phase separation taking place within a lamellar phase. This has been previously reported for TA16So8.15 Interestingly, it was also found that vesicles form spontaneously at room temperature and undergo a transition to large micelles at higher temperature, indicating that the solubility mismatch also affects the spontaneous curvature of the film.16 The main focus of this work is thus to evaluate the extent to which these self-assembly features are dependent on the chain length mismatch of the catanionic amphiphiles.15,20
Briefly, this paper is organized as follows. First, a global description of the phase behavior of the surfactant-water systems is presented, on the basis of comprehensive experimental data, followed by the main observations at low surfactant volume fraction (vesicle-micelle transitions). A theoretical model is employed in the Discussion to gain insight into the swelling behavior of some of the surfactants, together with a qualitative interpretation of the morphological transitions in the dilute regime.
2. Experimental section
Materials
Two series of alkyltrimethylammonium alkylsulfonates, (CH3)3N+(CH2)m-1CH3 SO3−(CH2)n-1CH3, referred to here as TAmSon, were prepared: (i) TA16Son series where n = 6–10, comprising then TA16So6, TA16So7, TA16So8, TA16So9 and TA16So10; (ii) a TAmSo8 series where m is 12, 14 and 16, comprising then TA12So8, TA14So8 and TA16So8 (common to both series).The parent surfactants hexadecyltrimethylammonium bromide (CTAB), tetradecyltrimethylammonium bromide (TTAB), dodecyltrimethylammonium bromide (DTAB) and sodium octylsulfonate (SOSo), were purchased from Sigma, while sodium hexylsulfonate (SHexSo), sodium heptylsulfonate (SHepSo), sodium nonylsulfonate (SNSo) and sodium decylsulfonate (SDeSo) were purchased from Fluka. The high purity chemicals were used as received.
The catanionic surfactant was prepared by precipitation at low temperature (ca. 4 °C) from equimolar micellar solutions (ca. 5 wt%) of the respective ionic surfactants. The catanionic precipitate was copiously washed with water and vacuum-dried, and then recrystallized 3–5 times to ensure that any salt and excess ionic surfactant are removed. Sodium atomic absortion indicates that these compounds are salt-free (typically ≪ 0.5 mol % NaBr) and further light microscopy and DSC data for the melting behavior, together with surface tension for cmc determination, do not show any meaningful evidence of impurities. Hence, this procedure is considered to yield salt-free catanionic surfactants with a high purity level.
Sample preparation and phase diagram determination
The samples used for the phase diagram mapping were prepared by weighing surfactant and water, followed by vigorous mixing. The samples were then put in a thermostated oven at the desired temperature. The phase boundaries of TA14So8 and TA12So8 on the dilute side with increasing temperature were determined by turbidity heating scans, further confirmed by samples at rest in the oven.Differential scanning calorimetry (DSC)
Heating-cooling-reheating scans were performed on a Setaram micro-calorimeter, at a fixed rate of 1.2 °C min−1. The standard cycle started with an isotherm of 1–4 h at 2 °C, in order to allow crystallization of the alkyl chains (especially critical in vesicle solutions due to the slow crystallization kinetics).Video-enhanced and polarized light microscopy (VEM and PLM)
A polarizing light microscope BX51 from Olympus, with differential interference contrast lenses, was used for phase assignment and visualization of aggregates. Images were acquired with an Olympus DP71 digital camera.Turbidity
Turbidity measurements were performed with a Cary 300 instrument equipped with a Peltier temperature control unit, at 400 nm and with a heating/cooling rate of 1 °C min−1. Hellma cuvettes with 0.1 cm optical path were used.Surface tension
Surface tension measurements in dilute solutions were performed with a DCAT21 tensiometer from DataPhysics (Wilhelmy plate method). The plate is made of a platinum-iridium alloy with roughed surfaces and precisely known geometry.Small-angle X-ray scattering (SAXS)
Synchrotron radiation SAXS was used to study the dilute lamellar phase from TA16So9, and intermediate phase from TA16So7, while conventional SAXS (Kratky camera) was performed on concentrated samples of all the surfactants. Synchrotron SAXS was performed at Max-Lab (Lund), at beam-line I711, with a wavelength of 1.1 Å and beam spot-size of approximately 79 μm. For data collection, a Mar165 area CCD detector from Mar Research was used with varying sample-to-detector distance. Data handling was performed with the Fit2D software application. The Kratky compact small-angle system was equipped with a position-sensitive detector (OED 50M from MBraun) containing 1024 channels of width 53.0 μm was used. The generator was a Seifert ID-300 X-ray, operating at 55 kV and 40 mA. A monochromator with a nickel filter was used to select the Cu-Kα radiation (l = 1.542 Å). The sample-to-detector distance was 277 mm.A lamellar liquid crystalline phase is characterized by a pattern of Bragg peaks with qn = q1n, with n = 1, 2, 3 (n is the nth Bragg reflection). The repeat distance d is obtained from the first Bragg peak according to:
 | (1) |
The q values for a hexagonal lattice are given by:
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√3:2:√7. Here d as given by eqn (1) is the distance between the planes containing rods, whereas the distance between rods is given by a. The rod radius, R, for the hexagonal phase and the bilayer thickness, δ, for the lamellar phase can then be obtained from eqn (3) and 4, respectively:
:√3:2:√7. Here d as given by eqn (1) is the distance between the planes containing rods, whereas the distance between rods is given by a. The rod radius, R, for the hexagonal phase and the bilayer thickness, δ, for the lamellar phase can then be obtained from eqn (3) and 4, respectively: | (3) |
| δ = Φd | (4) |
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, found for compound TA16So7, has Bragg peaks given by eqn (2) with the additional condition −h + k + l = 3n (with n integer), which limits possible reflections.39
m, found for compound TA16So7, has Bragg peaks given by eqn (2) with the additional condition −h + k + l = 3n (with n integer), which limits possible reflections.39
Small-angle neutron scattering (SANS)
The measurements were run in the SANSII instrument40 at SINQ lab in Paul Scherrer Institute (PSI), Switzerland. A range of scattering vectors q from 0.04 to 2.0 nm−1 was covered by a combination of two wavelengths λ and three sample-to-detector distances SDD in three different experimental set-ups (set 1: λ = 0.6 nm, SDD = 1.2 m; set 2: λ = 0.6 nm, SDD = 3 m; set 3: λ = 1.06 nm, SDD = 6 m). The wavelength resolution was 10%. A detector with 128 × 128 pixels and pixel size 0.44 cm was used. The samples were kept in 2 mm quartz cells (Hellma). The two-dimensional isotropic scattering curves have been corrected for detector efficiency by dividing the scattering data from the samples by the incoherent scattering data of neat water, and subsequently has been radially averaged and converted to absolute scale. These steps were performed using the BerSANS software.41 Background has then been subtracted, by subtracting the constant value of the incoherent scattering measured at high q values. The elongated micelles cross-section was determined with the SASfit software package (J. Kohlbrecher, PSI),42 for worm-like chains with excluded volume.433. Model calculations
As will be evident throughout this work, the key feature that controls the phase behavior of these compounds is the excess solubility of the anion ([Son−]) relative to that of the cation ([TAm+]). Modeling this effect, however, we assume that [Son−] is much larger than [TAm+], corresponding to essentially a salt free case. To rationalize the miscibility gap of the lamellar phase found for the weakly asymmetric catanionics, a previously developed modeling procedure was used.20 Briefly, the relevant forces for the stability of the lamellar phase are the attractive van der Waals force, the repulsive electrostatic force, and the short-range repulsive force.20 The van der Waals force is calculated according to44 | (5) |
 | (6) |
 | (7) |
 | (8) |
| σ = ½[Son−]effδ(Φ−1 − 1) | (9) |
Here [Son−]eff is the effective excess solubility of the short chain sulfonate ion relative to the TAm+ one, and this quantity is dependent on the concentration according to the charge regulation mechanism dictated by:20
 | (10) |
| Surfactant | T Kr /°C | cmc/mM |
|---|---|---|
| a At 40 °C, in mmol kg−1. | ||
| TA16So10 | 28 | 0.022a |
| TA16So9 | 16 | 0.042 |
| TA16So8 | 16 | 0.074 |
| TA16So7 | 17 | 0.15 |
| TA16So6 | 17 | 0.26 |
| TA14So8 | 19 | 0.21 |
| TA12So8 | 17 | 1.25 |
A further simplification considers these contributions to the total osmotic pressure to be additive, according to:
| Πtot = ΠvdW + ΠSRR + Πel | (11) |
Once the equation of state is calculated, the appearance of a van der Waals loop in the force curve implies a lamellar demixing. The phase limits can then be calculated by using a Maxwell construction, where the loop is divided in two areas of equal magnitude.15,20,44 The miscibility gap for compounds TA16So9 and TA16So10 were modeled using eqn (5)–(11), where the fitting parameters are the intrinsic solubility [So−]0 and the force amplitude A, which in combination influence the depth and height of the van der Waals loop and the limits of the gap.
4. Results
4.1 Binary surfactant-water phase diagrams
Prior to the phase behavior mapping, all the TAmSon catanionic surfactants prepared were checked for their solubility in water. Fig. 1a shows the DSC endothermic peaks associated with solubilization and from which the Krafft temperatures were obtained (in 0.5 wt% samples), as listed in Table 1.48Fig. 1b and Table 1 also show the cmc values from surface tension measurements. All the catanionic compounds studied here are soluble at room temperature, except TA16So10. For this compound, a 0.5 wt% solid dispersion only changes to a lamellar dispersion (L1 + Lα) at ca. 26 °C. Hence, it behaves similarly to the equal-chain length catanionics originally studied by Jokela et al.,8 which presented relatively high Krafft points. The cmc is seen to increase when the average pair chain length increases, as expected. The minimum area per molecule, as, is essentially the same for all compounds (≈0.60 nm2).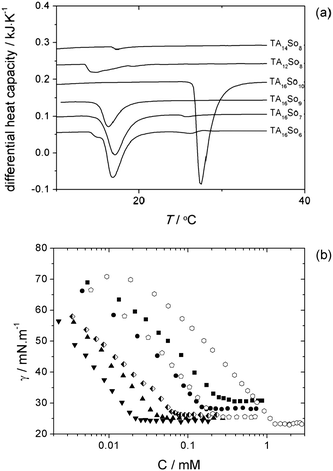 | ||
Fig. 1 Calorimetric and surface tension data for dilute solutions of the catanionic compounds: (a) DSC thermograms, vertically displaced for clarity, with the endothermic peaks signaling solubilization (0.5 wt% samples); (b) surface tension data, where filled symbols correspond to constant cationic chain length (TA16+), and open symbols to constant anionic chain length (So8−). Legend: TA16So6 (■); TA16So7 (●); TA16So9 (▲); TA16So10 (▼); TA14So8 (![[pentagon open]](https://www.rsc.org/images/entities/char_e1c1.gif) ) and TA12So8 ( ) and TA12So8 (![[hexagon open, flat-side down]](https://www.rsc.org/images/entities/char_e1c0.gif) ). TA16So8 is displayed with half filled diamonds. ). TA16So8 is displayed with half filled diamonds. | ||
The phase diagrams at 25 °C over the full concentration range for the binary surfactant-water systems are shown in Fig. 2. The uncertainty of the phase boundaries lies within ± 1 wt% for concentrations above 5 wt% surfactant (in most phase boundaries except those near the hydrated crystals) and ca. ± 0.2 wt% below 5 wt%. For TA16So10 the phase diagram was determined at 40 °C due to its higher Krafft point.
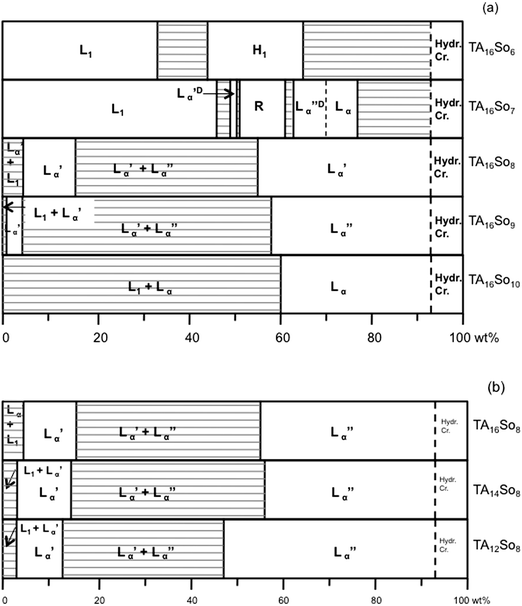 | ||
| Fig. 2 Isothermal phase diagrams of the binary surfactant-water systems at 25 °C. (a) TA16Son and (b) TAmSo8 series (TA16So10 at 40 °C). Legend: L1, micellar solution phase; H1, direct hexagonal liquid-crystalline phase; Lα, lamellar liquid-crystalline phase; Lα′, swollen lamellar liquid-crystalline phase; Lα′′, collapsed lamellar liquid-crystalline phase; Lα′D and Lα′′D, deffective lamellar phases; R, intermediate (rhombohedral) phase; hyd cryst., hydrated crystals. | ||
A global inspection of the phase diagrams allows us to divide the surfactants into 3 groups: (a) highly asymmetric compounds, comprising TA16So6 and TA16So7, which have highly charged films, lower Krafft points and whose behavior approaches that of a typical ionic micelle-forming surfactant; (b) weakly asymmetric compounds, comprising TA16So8, TA16So9, TA14So8, and TA12So8, which have weakly charged films and display swelling lamellar phases with a miscibility gap, i.e. coexistence of a concentrated (Lα′′) and a dilute (Lα′) lamellar phase; (c) approximately symmetric compounds, comprising TA16So10, which has essentially no charge, a higher Krafft point and a lamellar phase with very limited swelling (and no miscibility gap). These 3 groups also show differences in aggregation behavior in the dilute regime, where the weakly asymmetric amphiphiles display a temperature-dependent transition between vesicles and long micelles. A more detailed discussion of the phase behavior now follows, with the experimental evidence obtained.
 | ||
| Fig. 3 SAXS data for the TA16So6-water system at 60 wt% surfactant, with the SAXS pattern for a hexagonal phase with a rod radius R = 2 nm. The insert illustrates a phase-penetration scan where the surfactant concentration increases from left to right. A characteristic H1 birefringent texture is seen. | ||
In the TA16So7 case, the L1 phase is much wider, extending up to 45 wt%, and also less viscous than the TA16So6 one, which suggests a higher degree of micelle branching. With increasing concentration, instead of a hexagonal phase, bilayer-based phases now form, but with a peculiar and relatively complex sequence. First, a narrow defective lamellar phase Lα′D appears, followed by an intermediate phase and by another defective lamellar phase Lα′′D, that is practically juxtaposed to a normal lamellar phase Lα′′ (Fig. 4). The evidence for the defective lamellar structures comes from two main observations: first, there is a diffuse band at slightly lower q than the main Bragg peak in the SAXS 2-D and radially averaged patterns (Fig. 4a and c), signaling the presence of weakly correlated defects;29,49 secondly, non-ideal swelling is observed (Fig. 4d), also compatible with the presence of defects such as water-filled holes.50 The swelling becomes ideal only above 70 wt% (Lα′′ phase) and beyond this point, from fitting the model of classical one-dimensional swelling to the data we obtain δ = 2.7 nm (similar to TA16So8). In addition to the 1:2 Bragg peak periodicity, samples in the lamellar regions have a relatively low viscosity and myelinic figures appear on the water-rich side in a phase-penetration experiment (insert in Fig. 4d), consistent with a lamellar structure.51,52
 | ||
| Fig. 4
SAXS and PLM data from the TA16So7-water system at 25 °C. (a) Partially aligned 2-D scattering patterns: 50 and 66 wt%, Lα′D and Lα′′D phases, respectively, with a diffuse band clearly observed; 51 wt% marks the onset of the intermediate R phase, where by comparison with 50 wt%, the first peak (101 planes) is seen to develop from the diffuse band.49 (b) Typical PLM micrograph of the R phase. (c) Radially averaged scattering patterns for different samples. Full lines correspond to the section marked in (a) with a full line; the dashed line at 50 wt% is a horizontal section (marked in (a) with a dashed line) to highlight the diffuse band. The vertical lines indicate the expected q values for a rhombohedral Rm lattice. The assignment from left to right is (hkl) = (101), (012), (003), (110), (021), (104), (015) and (006). The 101 reflection arises from the diffuse band. (d) Swelling behavior in the 50–70 wt% region, with d-spacing vs. reciprocal volume fraction: filled triangles, Lα′D; filled circles, Lα′′D; open symbols, R phase (d003 spacing); pentagons, biphasic samples; squares, Lα (ideal swelling). Dashed line: ideal swelling fit yielding δ = 2.7 nm; solid line: polynomial fit to both Lα′D and Lα′′D; dotted line: polynomial fit to both Lα′′D and the R spacing. Insert: phase penetration scan showing the water-rich myelinic figures belonging to Lα′D. | ||
The intermediate phase (henceforth designated as R) appears in the 51–61 wt% range. It is highly viscous and shows some weak birefringence (Fig. 4b). The diffraction patterns for the whole composition range can be assigned to a rhombohedral lattice with space group Rm (cf. section 2 and Fig. 5). This space group has also been observed in the water/C16E6,39,53,54 and water/CTAB/3-sodium-2-hydroxy naphthoate (SHN)29,34 systems, where the structure was suggested to consist of planes of branched micelles in hexagonal arrangement, without interconnectivity between planes. These layers are arranged in a cubic-like ABC stacking, and besides elongated micelles, the planes can also be constituted by branched ribbons disposed in hexagonal arrangement or bilayers with perforations disposed also in hexagonal arrangement.54 In our case, the d003 periodicity—the main peak, analogous to that of the defective lamellar phases—lies in trend with the swelling of Lα′′D (and to some extent also to Lα′D), as can be seen in Fig. 4d. This suggests that the structure of the R phase is closely related to that of Lα′′D phase. This view is also supported by the fact that the 101 peak develops from the diffuse band49 when going from 50 to 51 wt% (Fig. 4a and c). When 55 and 58 wt% R samples are heated, the SAXS pattern of a defective lamellar phase is found around 50 °C (data not shown).
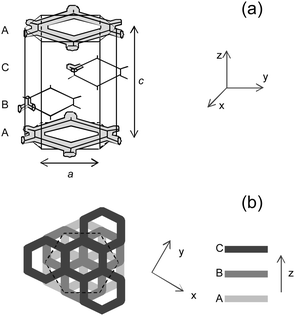 | ||
| Fig. 5 (a) Schematic three-dimensional representation of the intermediate phase (R m space group). A, B and C are similar layers, disposed in a ABC cubic-like stacking, and a and c are the unit cell dimensions as used in eqn (2). (b) Top-view of the intermediate phase for a better visualization of the ABC stacking. | ||
We note that the TA16So7 behavior in this region resembles that of the CTAB/SHN catanionic mixture,29,34 which forms an equilibrium intermediate phase between a defective (or random mesh) lamellar phase and a normal lamellar phase, at higher concentrations. There is also an analogy with the nonionic surfactants C16E6 (already mentioned),39,53,54 and C16E7,49 where a metastable intermediate phase was found on cooling a defective lamellar phase.
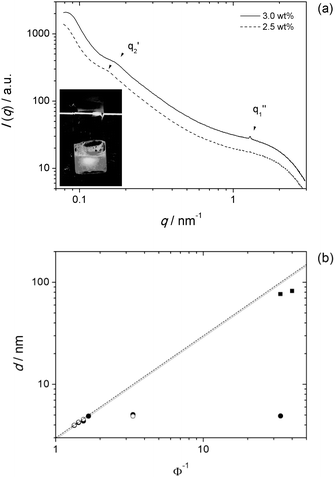 | ||
| Fig. 6 SAXS data for TA16So9 and TA16So10. (a) Scattering curves for 2.5 and 3.0 wt% TA16So9 samples showing the reflections for the swollen lamellar phase (at q2′) and the concentrated one (at q1′′). At 2.5 wt% the latter is absent. The insert shows the weak static birefringence observed for the 2.5 wt% sample. (b) Swelling curves for TA16So9 (full circles, Lα′′; squares, Lα′) at 25 °C and TA16So10 (empty circles) at 40 °C. Using ρ = 1, the data are fit to δ = 3.0 nm. From the biphasic samples at Φ−1 = 3.33, the limits of the collapsed phase are estimated at 59 wt% for TA16So9 and 60 wt% for TA16So10. | ||
The TA14So8 and TA12So8 compounds behave in a very similar way to TA16So8. Both display a miscibility gap in the lamellar phase region, with the swollen Lα′ showing a large stability range (unlike TA16So9). Samples in the miscibility gap have the typical milky-like appearance, whereas the single Lα′ is transparent, bluish and shows static birefringence. The Bragg peaks of the two coexisting lamellar phases are both clearly observed (Fig. 7a). Besides, the swelling behavior of both surfactants is ideally linear, apart from the miscibility gap region (Fig. 7b). Once again, assuming ρ = 1 for both compounds, the swelling curve is easily fitted to δ = 2.6 nm for TA14So8 and δ = 2.2 nm for TA12So8. From these values and the spacings of the two phases, the limits of the two-phase region are set to 15–56 wt% for TA14So8, and 13–46 wt% for TA12So8. The lower limit of the Lα′ is tentatively set at 1.8 wt% for both compounds, since the static birefringence vanishes below this composition.
 | ||
| Fig. 7 Swelling behavior for TA14So8 and TA12So8. (a) SANS curves for TA14So8 (solid line) and TA12So8 (dashed line), both at 25 wt% surfactant. The Bragg peaks of the swollen and collapsed lamellar phases are evident. (b) Swelling curve for both TA14So8 (open) and TA12So8 (solid symbols) systems, fit to δ = 2.6 (solid) and δ = 2.25 nm (dashed line) for TA14So8 and TA12So8 respectively. The discontinuity at Φ−1 = 4 is due to the coexistence of two lamellar spacings. This two-phase region corresponds to 14.5–55.7 wt% TA14So8 and 12.5–45.7 wt% TA12So8. | ||
4.2 Self-assembly at low concentration: elongated micelles and vesicles
We now focus our attention on the aggregation behavior at low concentrations, less than about 2 wt% surfactant. Broadly speaking one can see that: (i) the highly asymmetric compounds form elongated micelles and viscous solutions at relatively low concentrations, thus mimicking the classic behavior of single-chained ionic surfactants in the presence of salt or hydrotropes; (ii) the weakly asymmetric compounds form bilayer structures (vesicles) that change to elongated micelles only at high temperature; (c) symmetric compounds form bilayer dispersions that remain stable up to very high temperature.Fig. 8a shows SANS curves for TA16So6 (1 wt%), and TA16So7 (1 and 10 wt%). In the low q-part of the 1 wt% samples, the structure factor S(q) hides shape information and prevents a reliable fitting for all the q-range. Even so, neglecting the low-q part, one is able to fit the data to elongated micelles, with radius R = 2.0 nm,55 in agreement with the SAXS value for the hexagonal phase of TA16So6. The power law decay of q−1.7 observed slightly above q = 1 nm−1 indicates that these micelles are either wormlike and very flexible (with small persistence lengths lp)56 or highly branched. Visually, 1 wt% TA16So7 solutions are less viscous and flow more easily than 1 wt% TA16So6 solutions. These simple observations are consistent with the rheological experiments of Oda et al.,36 which showed that TA16So6 micelles go from wormlike to branched at about 3.8 wt%, while for TA16So7 this change over occurs at 0.71 wt% (hence, the much lower viscosity at 1 wt%). Temperature change has a weak effect on these samples (Fig. 8a).
 | ||
| Fig. 8 SANS data. (a) For TA16So6 1 wt% (squares), and TA16So7 1 wt% (circles) and 10 wt% (diamonds). The open symbols are for 25 °C and the solid ones are for 60 °C. The curves are shifted vertically for easier visualization. Ignoring the low-q part, dominated by the structure factor, both curves can be fit to an elongated micelle model with radius R = 2.0 nm (lines). (b) For TA16So9 0.5 wt% at different temperatures: 25 °C (□), 80 °C (○) and 95 °C (△). (c) For TA12So8 1 wt%, at different temperatures. | ||
With respect to the weakly asymmetric compounds (TA16So9, TA12So8 and TA14So8), an interesting observation is the spontaneous formation of vesicles found at low concentrations (<1.8 wt%), in all cases. For TA16So9, vesicles only form spontaneously by dissolution of the solid in water. In addition, the bluish solutions below 2 wt% (the lower limit of the Lα′ phase) slowly phase-separate with time, demonstrating that these vesicles are only mestastable. Fig. 8b displays SANS curves for 0.5 wt% samples. A q−2 power-law decay is observed, consistent with polydisperse vesicles and this slope remains constant with increasing temperature. Thus, no vesicle-to-micelle transition occurs for this surfactant, at least in the 25–95 °C range. With PLM, micrometre-sized vesicles are observed throughout.
In contrast, for TA14So8 and TA12So8 there is a transition into a colorless clear solution (L1 phase) with increasing temperature. At 25 °C and 80 °C SANS curves display a q−2 power-law decay for TA12So8, consistent with polydisperse vesicles (Fig. 8c). At 95 °C, the scattering intensity becomes much weaker and the power law decay shifts from q−2 to q−1, indicating a drastic structural change, likely a transition to relatively rigid elongated micelles (L1 phase).56
Just as with TA16So8, the TA14So8 and TA12So8 vesicles form spontaneously by a number of different pathways, namely (i) dissolution of the solid in water (Fig. 9a); (ii) cooling from the higher temperature L1 phase; and (iii) detachment of bilayer aggregates from the swelling dilute lamellar phase in excess solvent (Fig. 9b). Interestingly, similar myelinic figures have been observed by Buchanan et al. in similar phase-penetration experiments in water-nonionic52 and AOT-brine systems,51 however with the absence of bilayer fragment detachment.
 | ||
| Fig. 9 (a) Aged vesicle dispersion (ca. 1 month) of a TA12So8 1 wt% sample, obtained by dissolution of the solid in water. Similar micrographs for TA14So8 and TA16So9 dispersions were obtained. Scale bar: 50 μm. (b) Phase penetration scans for the vesicle-forming surfactants TA16So9, TA14So8 and TA12So8. Surfactant concentration increases from right to left. TA14So8 and TA12So8, but not TA16So9, show spontaneous detachment of bilayer fragments/vesicles (indicated by arrows). Scale bar (common to all images): 50 μm. | ||
In both TA14So8 and TA12So8 cases, the vesicular samples are bluish and translucent and neither phase separation nor flocculation are observed after several months at rest. Hence, a question remains whether such vesicles are thermodynamically stable, or metastable belonging instead to a two-phase Lα′ + L1 dispersion (like in TA16So9). This interesting and complex issue was left for further investigations, but we note that TA12So8 has been reported to form a stable vesicle phase in the presence of salt.57
The temperature-dependent phase behavior displayed by TA12So8 and TA14So8 is summed up in Fig. 10 where the structure evolution was determined through a combination of visual inspection, turbidity, PLM and SANS.58 Owing to the high stability over time displayed by the bluish samples at room temperature, we tentatively designated this region by Lves, noting that it may prove to be a metastable fine dispersion (i.e. effectively a L1 + Lα′ two-phase region).
 | ||
| Fig. 10 Self-assembled structures found at low concentration and as a function of temperature for the surfactants TA12So8 (dashed line), TA14So8 (small dashes) and TA16So8 (solid line):16 L1, micelles; Lves, vesicles. | ||
5. Discussion
5.1 Solubility mismatch and global phase behavior
The self-assembly behavior of the compounds herein studied can be broadly analyzed in terms of a charge regulation mechanism (cf.eqn (10)), the spontaneous curvature C0 of the film and the interplay of colloidal forces in the system. TA16So8 will be considered as the reference system15,16 and the discussion will be made in terms of an increase or decrease of the solubility mismatch relatively to this compound.Briefly, for TA16So8 the chain mismatch induces a certain charge density σ in the catanionic surface due to the fact that the short chain ion partitions into the bulk (eqn (9)). This charge density increases as the surfactant volume fraction Φs decreases.20 The observed lamellar-lamellar coexistence has been interpreted as resulting from a peculiar balance between repulsive forces—short-range entropic and electrostatic—and the attractive van der Waals forces.15 At high surfactant concentrations, σ is low and the swelling is controlled by the short-range repulsive force. Continuous addition of water will gradually remove So8− ions from the bilayers and increase σ. This effect strengthens the electrostatic force, which will stabilize the lamellar phase at higher dilutions. The sum of all these contributions to the total osmotic pressure can be plotted versus the interlamellar spacing L, as shown in Fig. 11a. There is a region in the curve where the net force is attractive and thus a van der Waals loop is originated, implying demixing of the lamellar phase into two different spacings. It is expected that high solubility mismatches will narrow the gap, while low mismatches will broaden it.
![(a) Contributions from the electrostatic repulsive force, the van der Waals atractive force (note the minus sign), and the short-range repulsive force to the total osmotic pressure, for the TA16So8 swelling. The shaded region is a Maxwell construction that divides the van der Waals loop in two equal areas, enabling the determination of the miscibility gap limits. The inserts are a schematic representation of the collapsed (left) and swollen lamellar (right) phases. (b) Total osmotic pressure curves for the swelling compounds, with [Son−]0 = 2.5 mM describing well the phase behavior for TA16So8, [Son−]0 = 0.3 mM for TA16So9, and [Son−]0 = 0.04 mM for TA16So10.](/image/article/2011/SM/c0sm00477d/c0sm00477d-f11.gif) | ||
| Fig. 11 (a) Contributions from the electrostatic repulsive force, the van der Waals atractive force (note the minus sign), and the short-range repulsive force to the total osmotic pressure, for the TA16So8 swelling. The shaded region is a Maxwell construction that divides the van der Waals loop in two equal areas, enabling the determination of the miscibility gap limits. The inserts are a schematic representation of the collapsed (left) and swollen lamellar (right) phases. (b) Total osmotic pressure curves for the swelling compounds, with [Son−]0 = 2.5 mM describing well the phase behavior for TA16So8, [Son−]0 = 0.3 mM for TA16So9, and [Son−]0 = 0.04 mM for TA16So10. | ||
For the TA16Son series, when the solubility mismatch is decreased by going from So8− to So9−, the stability of the swollen lamellar Lα′ phase is greatly reduced (its upper limit shifts from 15 to 3 wt%). C0 does not seem to be significantly altered but the decrease in σ weakens the electrostatic repulsive force that stabilizes Lα′. Applying the charge-regulation model to TA16So9 (cf. section 3), the miscibility gap is nicely fitted to an intrinsic solubility for So9−, [So9−]0, of 0.3 mM (Fig. 11b and Table 2).
As the mismatch is further lowered in So10−, σ is now too low to be able to stabilize the swollen Lα′ at any point, hence this phase vanishes and only the collapsed lamellar forms. In fact, the excess intrinsic solubility of So10− can be estimated by assuming a logarithmic dependence of the intrinsic solubility with chain length (in analogy with the cmc dependence on chain length), using the determined values for So8− and So9. Hence, we extrapolate [So10−]0 = 0.04 mM and from the equation of state we get an upper boundary of the nominal Lα′ phase at 0.1 wt%. At this concentration we observe macroscopic phase separation, with the collapsed lamellar phase creaming to the top of the vial, and an isotropic solution filling most of the sample volume. Hence, for TA16So10 the swollen lamellar phase is absent. These results show that the charge regulation model is able to capture the miscibility gap dependence on solubility mismatch.20
If the short chain solubility is now raised from So8− to So7− or So6−, a boost in the surface charge density σ for an identical surfactant concentration is induced. This effect not only strengthens the electrostatic force, but it also increases the spontaneous curvature C0 of the surfactant film, so that the swollen Lα′ phase is replaced with a large L1 region for both So6− and So7−. At higher concentrations, defective bilayer-based morphologies are found for So7−, all with C0 lying somewhere between that of rods and normal bilayers, again demonstrating that the spontaneous curvature increases with charge density59 (and therefore with the chain-length asymmetry). Switching to So6− the even higher C0 now favors only the rod-based hexagonal phase.
Considering now the TAmSo8 compounds, the solubility mismatch is gradually lowered by going from m = 16 to 14 and 12, and the changes in phase behavior are less pronounced than in the TA16Son series. The lamellar miscibility gap is maintained in the series. The upper limit of the swollen Lα′ lowers for TA14+ and TA12+, as expected if σ is decreased. However, the stability range of Lα′ for these compounds is higher than that of TA16So9. This could be explained in part by the fact that changing the solubility of the less soluble ion always has a smaller impact on σ than changing the solubility of the most soluble one. In addition, the reduction in bilayer thickness δ—2.6 nm for TA14So8 and 2.2 nm for TA12So8—can induce (or strengthen) undulation repulsive forces, and simultaneously lower the magnitude of the van der Waals attraction. Both these effects would stabilize Lα′ further and to a similar extent as in TA16So8. Support for the role of undulations comes from the comparison of TA12So8 with the dodecyltrimethylammonium chloride/sodium octylsulfonate (DTAC/SOSo) system, studied by Coldren et al.57 In this salt-containing mixture, only one lamellar phase Lα is found, as one would expect if salt screens the electrostatic force and repulsive undulation forces predominate.
5.2 Aggregation in the dilute region
TA16So8 shows a vesicle-to-micelle transition that depends both on concentration and temperature, below 2 wt%.16 The fact that this transition is temperature-driven can be explained, at least partly, by the fact that solubility usually increases with temperature, and thus an increase in T induces a net increase in solubility mismatch. The increase in charge density, σ, should then increase C0 up to a point where the formation of large micelles is now preferred. Bearing in mind the charge-regulation model,31 lowering the concentration should also lower the temperature required for such a transition to occur, thus explaining the inclination of the Lves/L1 boundary.The temperature effect on the solubility of So8− is equivalent to a replacement of this ion with So7− or So6− at constant T and concentration. Since a higher fraction of short chain ions dissolve in the bulk, both σ and C0 increase favoring the formation of L1 micellar phases to the detriment of bilayers. In the opposite direction, decreasing the short chain solubility with So9− and So10− inhibits the transition to the L1 phase even at temperatures as high as 95 °C. Clearly, σ is not high enough to induce a transition to micelles, even at such high temperatures.
Conversely, if So8− is kept fixed, the TA+ solubility is increased on going from TA16+ to TA14+ and to TA12+, hence the solubility mismatch decreases. However, this effect seems not to be as pronounced as in the previous series, since the transition to a single L1 phase is observed in all cases. Still, the transition temperature for TA14So8 and TA12So8 is higher than for TA16So8. Furthermore, the transition in TA12So8 is almost independent of concentration, as one would expect if the mismatch effect is weaker.
These considerations are further supported by a comparison of our results with the literature on the aqueous behavior of CTAB in the presence of small ions. Because Br− is much more soluble than So6−, a higher σ favors spherical micelles up to 14 wt% surfactant.60 Replacing Br− with the less soluble salicylate ion induces micelle growth at much lower concentrations,37,61 in analogy with So7−. If the solubility is even further decreased with the 3-hydroxy-naphthalene-2-carboxylate ion, vesicles are observed.32,35,62–64 Here, a temperature-driven vesicle-to-micelle transition is also observed, as is the case of TA16So8.63
Finally, we note that the self-assembly trends described here, both at high and low concentration, should be common to other families of salt-free catanionics of the Cm+Cn− type, with the influence of chain length/solubility mismatch being slightly modulated by the details of headgroup composition.
6. Conclusions
We have demonstrated that a change in the relative solubility of the amphiphilic ions of salt-free catanionic surfactants, has profound effects on the aqueous phase behavior. Low solubility mismatch controls mainly the electrostatic repulsive force (through σ) between lamellae, being crucial for the stability of lamellar phases and the existence of miscibility gaps. As the solubility mismatch increases, it helps stabilize the dilute lamellar phases, reducing the miscibility gap. For high solubility mismatch, the spontaneous curvature C0 is also significantly affected due to the large increase in σ. This leads to the formation of elongated micelles in the dilute side, and hexagonal/intermediate phases in the concentrated one. For fixed molecular structure (TAm+Son−), increasing the temperature also changes the solubility mismatch, and a vesicle-to-micelle transition is found for some compounds.Finally, besides the solubility mismatch, the overall chain length of the catanionic compound also plays an important role, since it affects undulation and van der Waals forces (through the bilayer thickness), which are important to stabilize different aggregates and liquid-crystalline phases.
Acknowledgements
We thank Prof. V. A. Raghunathan for providing us detailed information on the intermediate phase of the CTAB/SHN system. We are grateful for financial support from CIQ-UP, Porto University, and from the Swedish Research Council (VR). BFBS is grateful to FCT for the PhD grant SFRH/BD/24966/2005 and to Calouste Gulbenkian Foundation through grant 107530. Part of this work was performed at the Swiss spallation neutron source SINQ, Paul Scherrer Institute, Villigen and thanks are due to our local contact there, Sandor Balog. This work has also been partly supported by the European Commission under the 6th Framework Programme through the Key Action: Strengthening the European Research Area, Research Infrastructures. Contract no: RII3-CT-2003-505925. Max-Lab, Lund, is acknowledged for providing us beam-time, as well as technical assistance.References and notes
- K. A. Bartscherer, M. Minier and H. Renon, Fluid Phase Equilib., 1995, 107, 93–150 CrossRef CAS.
- R. Skurtveit and U. Olsson, J. Phys. Chem., 1992, 96, 8640–8646 CrossRef CAS.
- J. F. Rusling and H. Zhang, Langmuir, 1991, 7, 1791–1796 CrossRef CAS.
- J. E. Klijn and J. B. F. N. Engberts, J. Am. Chem. Soc., 2003, 125, 1825–1833 CrossRef CAS.
- M. Rosa, M. G. Miguel and B. Lindman, J. Colloid Interface Sci., 2007, 312, 87–97 CrossRef CAS.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- J. C. Hao and H. Hoffmann, Curr. Opin. Colloid Interface Sci., 2004, 9, 279–293 CrossRef CAS.
- P. Jokela, B. Jönsson and A. Khan, J. Phys. Chem., 1987, 91, 3291–3298 CrossRef CAS.
- A. Khan and E. F. Marques, Curr. Opin. Colloid Interface Sci., 2000, 4, 4002–4410.
- C. Tondre and C. Caillet, Adv. Colloid Interface Sci., 2001, 93, 115–134 CrossRef CAS.
- T. Zemb and M. Dubois, Aust. J. Chem., 2003, 56, 971–979 CrossRef CAS.
- M. Dubois, B. Demé, T. Gulik-Kryzwicki, J.-C. Dedeiu, C. Vautrin, S. Désert, E. Perez and T. Zemb, Nature, 2001, 411, 672–675 CrossRef CAS.
- H. Li and J. Hao, J. Phys. Chem. B, 2008, 112, 10497–10508 CrossRef CAS.
- E. Maurer, L. Belloni, T. Zemb and D. Carrière, Langmuir, 2007, 23, 6554 CrossRef CAS.
- B. F. B. Silva, E. F. Marques and U. Olsson, J. Phys. Chem. B, 2007, 111, 13520–13526 CrossRef CAS.
- B. F. B. Silva, E. F. Marques and U. Olsson, Langmuir, 2008, 24, 10746–10754 CrossRef CAS.
- T. Zemb, M. Dubois, B. Demé and T. Gulik-Kryzwicki, Science, 1999, 283, 816–819 CrossRef CAS.
- K. L. Herrington, E. W. Kaler, D. D. Miller, J. A. Zasadzinski and S. Chiruvolu, J. Phys. Chem., 1993, 97, 13792–13802 CrossRef CAS.
- B. F. B. Silva and E. F. Marques, J. Colloid Interface Sci., 2005, 290, 267–274 CrossRef CAS.
- B. F. B. Silva, E. F. Marques, U. Olsson and P. Linse, J. Phys. Chem. B, 2009, 113, 10230–10239 CrossRef CAS.
- H. Edlund, A. Sadaghiani and A. Khan, Langmuir, 1997, 13, 4953–4963 CrossRef CAS.
- A. González-Pérez, M. Schmutz, G. Waton, M. J. Romero and M. P. Krafft, J. Am. Chem. Soc., 2007, 129, 756–757 CrossRef CAS.
- Y. W. Shen, J. C. Hao and H. Hoffmann, Soft Matter, 2007, 3, 1407–1412 RSC.
- A. Song, S. Dong, X. Jia, J. Hao, W. Liu and T. Liu, Angew. Chem., Int. Ed., 2005, 44, 4018–4021 CrossRef CAS.
- G. Persson, H. Edlund, E. Hedenström and G. Lindblom, Langmuir, 2004, 20, 1168–1179 CrossRef CAS.
- J. Eastoe, P. Rogueda, D. Shariatmadari and R. Heenan, Colloids Surf., A, 1996, 117, 215–225 CrossRef CAS.
- K. Bijma and J. B. F. N. Engberts, Langmuir, 1997, 13, 4843–4849 CrossRef CAS.
- R. T. Buwalda, M. C. A. Stuart and J. B. F. N. Engberts, Langmuir, 2000, 16, 6780–6786 CrossRef CAS.
- S. K. Ghosh, R. Ganapathy, R. Krishnaswamy, J. Bellare, V. A. Raghunathan and A. K. Sood, Langmuir, 2007, 23, 3606–3614 CrossRef CAS.
- S. K. Ghosh and V. A. Raghunathan, Langmuir, 2009, 25, 2622–2628 CrossRef CAS.
- P. A. Hassan, S. J. Candau, F. Kern and C. Manohar, Langmuir, 1998, 14, 6025–6029 CrossRef CAS.
- P. A. Hassan, J. Narayanan, S. V. G. Menon, R. A. Salkar, S. D. Samant and C. Manohar, Colloids Surf., A, 1996, 117, 89–94 CrossRef CAS.
- T. K. Hodgdon and E. W. Kaler, Curr. Opin. Colloid Interface Sci., 2007, 12, 121–128 CrossRef CAS.
- R. Krishnaswamy, S. K. Ghosh, S. Lakshmanan, V. A. Raghunathan and A. K. Sood, Langmuir, 2005, 21, 10439–10443 CrossRef CAS.
- E. Mendes, R. Oda, C. Manohar and J. Narayanan, J. Phys. Chem. B, 1998, 102, 338–343 CrossRef CAS.
- R. Oda, J. Narayanan, P. A. Hassan, C. Manohar, R. A. Salkar, F. Kern and S. J. Candau, Langmuir, 1998, 14, 4364–4372 CrossRef CAS.
- U. Olsson, O. Söderman and P. Guéring, J. Phys. Chem., 1986, 90, 5223–5232 CrossRef CAS.
- B. F. B. Silva, E. F. Marques, U. Olsson and R. Pons, Langmuir, 2010, 26, 3058–3066 CrossRef CAS.
- S. S. Funari and G. Rapp, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7756–7759 CrossRef CAS.
- P. Strunz, K. Mortensen and S. Janssen, Phys. B, 2004, 350, e783–e786 CrossRef CAS.
- U. Keiderling, Appl. Phys. A: Mater. Sci. Process., 2002, 74, S1455–S1457 CrossRef CAS.
- J. Kohlbrecher, SASfit ver. 0.86, available at: http://kur.web.psi.ch/sans1/SANSSoft/sasfit.html.
- J. S. Pedersen and P. Schurtenberger, Macromolecules, 1996, 29, 7602–7612 CrossRef CAS.
- H. Wennerström, Langmuir, 1990, 6, 834–838 CrossRef.
- D. F. Evans and H. Wennerström, The Colloidal Domain: Where Physics, Chemistry, Biology and Technology Meet, 2nd edn, John Wiley & Sons, Inc, New York, 1999 Search PubMed.
- S. Engström and H. Wennerström, J. Phys. Chem., 1978, 82, 2711 CrossRef CAS.
- The surface potential is the difference in electrical potential between the surface and the midplane, where the potential is set to zero by convention. The use of the modulus enables the use of eqn (10) for positive and negative counterions or surface charge densities, provided that the counterions are monovalent.
- Since crystallization of bilayer alkyl-chains in vesicles has a complex kinetic behavior, an evaluation of the transitions enthalpies changes has not been carried out.
- M. Imai, A. Kawaguchi, A. Saeki, K. Nakaya, T. Kato, K. Ito and Y. Amemiya, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 6865–6874 CrossRef CAS.
- J. Gustafsson, G. Orädd, G. Lindblom, U. Olsson and M. Almgren, Langmuir, 1997, 13, 852–860 CrossRef CAS.
- M. Buchanan, J. Arrault and M. E. Cates, Langmuir, 1998, 14, 7371–7377 CrossRef CAS.
- M. Buchanan, S. U. Egelhaaf and M. E. Cates, Langmuir, 2000, 16, 3718–3726 CrossRef CAS.
- C. E. Fairhurst, M. C. Holmes and M. S. Leaver, Langmuir, 1997, 13, 4964–4975 CrossRef CAS.
- M. Leaver, A. Fogden, M. Holmes and C. Fairhurst, Langmuir, 2001, 17, 35–46 CrossRef CAS.
- For the fitting analysis, excluding the low-q part, the contour and Kuhn lengths can take virtually any value, as long as the former is higher than the latter. Therefore, the only parameter with physical meaning that can be extracted is the micelle radius. The fitting can be run automatically provided that the contour length is larger than the Kuhn length.
- G. Porte, in Neutrons, X-Rays and Light: Scattering Methods Applied to Soft Condensed Matter, ed. P. Lindner and T. Zemb, Elsevier Science B.V., Amsterdam, 1st edn, 2002, pp. 299–316 Search PubMed.
- B. Coldren, H. Warriner, R. Van Zanten, J. A. Zasadzinski and E. B. Sirota, Langmuir, 2006, 22, 2474–2481 CrossRef CAS.
- It is worth mentioning that there is a discrepancy between the temperature of the phase transitions in the samples used for the phase diagrams and the ones used in SANS. This is because the latter were prepared in D2O, which shifts the phase boundaries in more than 10 °C.
- A. Fogden and J. Daicic, Colloids Surf., A, 1997, 129–130, 157–165 CrossRef.
- L. Coppola, R. Gianferri, I. Nicotera, C. Oliviero and G. A. Ranieri, Phys. Chem. Chem. Phys., 2004, 6, 2364–2372 RSC.
- V. K. Aswal, P. S. Goyal and P. Thiyagarajan, J. Phys. Chem. B, 1998, 102, 2469–2473 CrossRef CAS.
- P. A. Hassan, B. S. Valaulikar, C. Manohar, F. Kern, L. Bourdieu and S. J. Candau, Langmuir, 1996, 12, 4350–4357 CrossRef CAS.
- S. V. G. Menon, C. Manohar and F. Lequeux, Chem. Phys. Lett., 1996, 263, 727–732 CrossRef CAS.
- E. Mendes, J. Narayanan, R. Oda, F. Kern, S. J. Candau and C. Manohar, J. Phys. Chem. B, 1997, 101, 2256–2258 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |