Polymer films prepared using ionically crosslinked soft core–shell nanoparticles: a new class of nanostructured ionomers†
Orawan
Pinprayoon
a,
Robert
Groves
b,
Peter A.
Lovell
a,
Somjit
Tungchaiwattana
a and
Brian R.
Saunders
*a
aPolymer Science and Technology Group, The School of Materials, The University of Manchester, Grosvenor Street, M1 7HS, UK. E-mail: Brian.saunders@manchester.ac.uk
bSynthomer Ltd., Temple Fields, Harlow, Essex CM20 2BH, UK
First published on 15th October 2010
Abstract
Although conventional ionomers have been studied for more than 40 years, reports of ionomers prepared using aqueous core–shell nanoparticle dispersions are lacking. In this study we examine the structure and properties of new soft core–shell elastomeric nanoparticles and ionomer films. The nanoparticles consist of a soft poly(Bd) (Bd is 1,3-butadiene) core and a poly(Bd-co-MAA) shell (MAA is methacrylic acid). The nanoparticles were prepared by sequential emulsion polymerisation and were characterised using photon correlation spectroscopy, TEM, PSDA (particle size distribution analysis) and potentiometric titration. Robust ionomer films were formed by casting mixed core–shell nanoparticle/ZnO dispersions at room temperature. The ZnO particles provided aqueous Zn2+ which neutralised the carboxylate groups and imparted ionic crosslinking. The mechanical properties of the films were investigated using DMTA (dynamic mechanical thermal analysis) and stress–strain measurements. Neutralisation substantially increased the film modulus values. This was ascribed to the formation of a poly(Bd-co-MAA)/Zn2+-rich layer that moved inwards from the periphery of the nanoparticles as the degree of neutralisation increased. In contrast to conventional ionomers, the mechanical properties of the present films depend on ionic crosslinking within a honeycomb-type continuous phase that percolates the film. The storage and tensile moduli for the films increased linearly with neutralisation. The films studied here are a new class of nanostructured ionomers and the results obtained should be generally applicable to other films comprising soft-core–shell nanoparticles containing surface carboxylate groups.
1. Introduction
Core–shell nanoparticles have well-defined and controllable structures and have attracted considerable fundamental interest.1–9 Stable colloidal dispersions of such particles have been prepared using copolymer micelles,5 heteroaggregation4 and sequential seeded emulsion polymerisation.1,3 The latter method is relatively easy to control and has the greatest potential for application since it can be cost-effectively scaled up.10 Because of the ability to exert fine control over the structure of core–shell nanoparticles they are appealing for fundamental studies and for manipulation of end-use properties. Core–shell nanoparticles with a soft core and a crosslinked shell11 are particularly interesting because they provide elastomeric films with tuneable mechanical properties.2 The present study aims to demonstrate how the nanometre scale structure of soft elastomeric nanoparticles can be used to tune the mechanical properties of films cast from their dispersions. The films studied in this work have elastic moduli values that increase substantially due to ionic crosslinking. They are structurally different to films prepared using conventional latexes12,13 and also conventional ionomers.14 In this study we test our potentially useful hypothesis that the mechanical properties of films formed from soft core–shell nanoparticles can be controlled principally by the ionic crosslink density within the shell of the nanoparticles.In this study soft core–shell nanoparticles comprising phases produced from Bd and MAA (1,3-butadiene and methacrylic acid) have been prepared and studied for the first time. The films prepared from these nanoparticles rely on ionic crosslinking for enhancement of their mechanical properties. Although poly(Bd)-containing core–shell particles prepared using copolymer micelles have been reported elsewhere,6 there have been very few reported studies involving poly(Bd)-based core–shell nanoparticles prepared using emulsion polymerisation.15,16Nanoparticles containing a soft poly(Bd) core should be ductile and have potential for subsequent covalent crosslinking. This opens up potential application as surface coatings, pressure sensitive adhesives and also as biomaterials.17
An ionomer is a polymer having a relatively small percentage of ionic groups18 (typically up to 10–15 mol%). Conventional ionomers have been known for more than 40 years and seminal studies of ionomers were performed by Eisenberg et al.14,19,20 Many of the principles governing their structures and mechanical properties are well understood.19,20 Conventional ionomers are usually prepared using solutionpolymerisation, casting or melt processing and have included Bd-containing systems.21,22 Most of the knowledge concerning ionomers originates from the study of random ionomers, e.g., based on lightly crosslinked polystyrene.23Ionomers continue to attract considerable interest.24 We recently reported work involving a different approach to ionomer preparation which involved mixingdispersions of poly(Bd-co-MAA) nanoparticles and ZnO particles,25 the latter acting as a source of Zn2+ for the ionic crosslinking of the carboxylate groups from MAA repeat units. The nanoparticles were prepared using monomer-flooded batch emulsion copolymerisations26 and so the radial distribution of MAA repeat units was not controlled and was diffuse. Those poly(Bd-co-MAA) nanoparticles are termed conventionalnanoparticles for this study. The ionic crosslinking in films cast from those systems was shown to occur mostly in the periphery of the particles, suggesting that only MAA repeat units in the outer regions were involved in crosslinking.
In the present work the crosslinking and mechanical properties of films cast from soft core–shell nanoparticles prepared using ‘seed-and-grow’ semi-batch emulsion copolymerisation are investigated. The present nanoparticles have a much improved separation between core and shell, consisting of a poly(Bd) core and a poly(Bd-co-MAA) shell. Through structuring of the particles in this way, the ionic crosslinking is forced to occur only in the particle shell regions to give films with a honeycomb-type film morphology. The general approach is shown in Scheme 1. This is very different to the morphology of conventional ionomer materials considered elsewhere,18–20,23 which is homogeneous on the scale of ca. 100 nm. It is also different to the conventional polystyrene latex ionomers investigated by Weiss et al.27–29 which were not core–shell particles.
 | ||
| Scheme 1 Preparation of nanostructured ionomer films composed of ionically crosslinked soft core–shell nanoparticles. | ||
Here, we compare the properties of three new core–shell nanoparticle dispersions and films to those of the previous, conventional, poly(Bd-co-MAA) nanoparticle system and also more generally to conventional ionomers. It is shown for the first time that deliberate confinement of the MAA repeat units to an outer shell yields new polymer ionomer films with mechanical properties controlled by the shell composition. The overall consequence of confining the ionic crosslinking to well-defined shells is to provide the opportunity, for the first time, of tuning the mechanical properties of these nanostructured ionomer films through nanoparticle design. The method used to prepare the films is versatile and straightforward. The method and results presented should be applicable to any type of water-dispersible soft core–shell nanoparticles where the shell contains carboxylic acid groups.
2. Experimental
2.1. Reagents and nanoparticle preparation
All reagents and materials were used as received and were provided by Synthomer Ltd. Poly(Bd-co-MAA) dispersions were prepared using either batch emulsion polymerisation or a sequential (two-stage) emulsion polymerisation. The code CxS is used in this study to identify the core–shell nanoparticle types, whereby x represents the nominal core volume fraction in the nanoparticle. For example, C75S had a nominal core volume fraction (ϕc) of 0.75 and a nominal shell volume fraction (ϕsh) of 0.25. The core-only latex is C100. Additionally, a ‘shell-only’ poly(Bd-co-MAA) latex (S100) was prepared directly without use of a poly(Bd) core.S100 nanoparticles were prepared by monomer-flooded batch emulsion copolymerisation of a monomer mixture containing 94 wt% of Bd and 6 wt% of MAA using the procedure described in detail elsewhere.26 The core particles (C100) were prepared using a similar procedure as described for S100 with the exception that MAA was omitted.
The method used for preparing C25S is described below and is representative of all the semi-batch emulsion copolymerisations used here. The only differences for C50S and C75S were the proportions of monomer used in the two stages. A solution containing water (1210 ml), ethylenediaminetetraacetic acid (3.63 g), hydroxymethanesulfinic acid monosodium salt dihydrate (0.36 g), and poly(Bd) C100 seed (600 g, 40 wt%) was mixed and added to the reactor. Two feed mixtures were prepared. Feed 1 was a solution containing Bd (668 g), MAA (58 g), tert-butyl hydroperoxide (t-BHP, 1.56 g) and t-dodecyl mercaptan (7.26 g). Feed 2 comprised 30 wt% aqueous NaDBS (15.4 g) diluted with water (565 ml). The polymerisation was performed at 60 °C and was initiated by an injection of a solution of t-BHP (0.52 g) in water (21.61 g), which was followed by metering of Feed 1 and Feed 2 under nitrogen such that each feed was complete after 360 min. The reaction was allowed to proceed until a conversion of at least 85% was obtained. Table 1 shows a summary of the compositions used to prepare the nanoparticle dispersions investigated here.
| Core/shell | Code | ϕ c(nom) a | ϕ c(act) b | x MAA(sh) c | d h d/nm | d (PSDA) e/nm | d ( TEM ) f/nm | d (TEM)cal g/nm | N p(end) /Np(init)h |
|---|---|---|---|---|---|---|---|---|---|
| a Nominal volume fractions of core present in the nanoparticles based on 100% conversion. b Actual volume fractions of core present in the nanoparticles based on conversions. c Nominal values for the mole fraction of MAA in the shell based on the compositions used to prepare the nanoparticles. d Hydrodynamic diameter measured using PCS. e Number-average diameter from PSDA measurements. f Number-average nanoparticle diameter from TEM data. The number in brackets is the coefficient of variation. g Calculated number-average diameter using the number of core particles present at the start of shell growth and the mass of monomer mixture converted during shell growth. h Ratio of the total number of particles in the reactor at the end of shell growth, Np(end), to its initial value, Np(init), calculated from the nanoparticle dispersion solid contents and d(TEM) values assuming the density of all phases in the particles to be 1.0 g cm−3. The Np(init) values for C75S, C50S and C25S were 1.0 × 1019, 7.2 × 1018 and 2.8 × 1018, respectively. | |||||||||
| Poly(Bd) | C100 | 1.00 | 1.00 | — | 94 | 68 | 55 [38%] | — | — |
| Poly(Bd)/poly(Bd-co-MAA) | C75S | 0.75 | 0.765 | 0.165 | 101 | 68 | 57 [30%] | 60 | 1.2 |
| Poly(Bd)/poly(Bd-co-MAA) | C50S | 0.50 | 0.509 | 0.079 | 109 | 76 | 69 [28%] | 69 | 1.0 |
| Poly(Bd)/poly(Bd-co-MAA) | C25S | 0.25 | 0.273 | 0.052 | 117 | 93 | 85 [22%] | 82 | 0.9 |
| Poly(Bd-co-MAA) | S100 | — | — | — | 99 | 83 | 76 [22%] | — | — |
2.2 Nanoparticle film preparation
The method used to prepare the nanoparticle films involved casting colloidally stable aqueous dispersions25 containing the CxS nanoparticles and a source of Zn2+. A ZnO particle dispersion (Aquaspersions Ltd., UK, particle size 2 µm) was used for the latter and this was added to the nanoparticle dispersion maintained at a pH of 8.0 by pre-addition of aqueous KOH solution. Both K+ and Zn2+ ions were incorporated into the nanoparticles; however, the Zn2+ ions were in excess for the majority of the neutralised films considered. The nominal percentage neutralisation (α) used here is the molar % of total cation added with respect to the nominal moles of MAA groups present. The KOH added corresponded to 15 mol% neutralisation for the S100 nanoparticles and 25 mol% neutralisation for all of the CxS nanoparticle systems. Nanoparticle films were cast onto clean glass plates and dried at 25 °C in a humidity-controlled environment (50% RH). The total solids content for the dispersions used at the point of film preparation was 30 wt%. The dried films had an average thickness in the range of 200–400 µm. For the aging study, the antioxidant Wingstay L (Eliochem) was added to the dispersions at a level of 0.6 wt% to polymer prior to film casting (see Section 3.3).2.3 Physical measurements
Potentiometric titrations were conducted using a Mettler DL53 autotitrator. Photon correlation spectroscopy (PCS) measurements were performed using a BI-9000 Brookhaven Instrument Corporation light scattering apparatus fitted with a 20 mW He–Ne laser and a photomultiplier detector set at a 90° scattering angle. Transmission electron microscopy (TEM) measurements were performed using a Philips CEM 200 operating at 200 keV. The nanoparticles were negatively stained using a 2% phosphotungstic acid solution before being deposited on a Holey Carbon grid (Agar Scientific Ltd.) and dried at room temperature overnight. Number-average particle sizes were determined by measuring at least 300 particles. Particle size distribution analysis (PSDA) was used for measuring the particle size and size distribution of dispersed particles using a PL-PSDA (Polymer Laboratories) instrument. Attenuated total reflectance Fourier-transform infrared (FTIR) measurements were conducted on a Nicolet 5700 FTIR instrument. Atomic force microscopy (AFM) measurements were performed using a Veeco CP-IIscanning probe microscope equipped with a silicon nitride cantilever using the tapping and phase contrast modes at ambient conditions. Freeze-fractured cross-sections for TEM and AFM were prepared by liquid nitrogen immersion of films. Dynamic mechanical thermal analysis (DMTA) was performed in tensile mode at 1 Hz on a TA-Q800 Instrument DMA which provided storage modulus (E′), loss modulus (E″) and tan δ values (=E″/E′). Stress–strain measurements were conducted at 25 °C, 50% RH using a Hounsfield tensile testing instrument equipped with a laser extensometer. Films were cut into dumb-bell shaped specimens with overall length 75 mm and constricted width of 4.0 mm. The extension rate was 500 mm min−1.3. Results and discussion
3.1 Structure and composition of poly(Bd-co-MAA) core–shell nanoparticles and films
Three core–shell poly(Bd)/poly(Bd-co-MAA) nanoparticle latexes were investigated (see Table 1). Conventional poly(Bd-co-MAA) nanoparticle dispersion (S100) and poly(Bd) nanoparticle dispersion (C100) were used for comparison. The CxS and S100 nanoparticles had the same overall nominal MAA mole fractions (xMAA) of 0.04 on a whole particle basis. Fig. 1(a) shows the growth of hydrodynamic particle diameter (from PCS) with feed time for the C25S/C75S dispersions and demonstrates the expected trends. PSDA was used to measure the size distribution of the final dispersions. The data are shown in Fig. 1(b) (and Table 1). Although the data for C100 and C75S are similar, it can be seen that the whole particle size distribution moves to larger diameter values as ϕc decreases (and ϕsh increases). This trend is also evident from comparison of the particle size data obtained from TEM (Table 1). The larger number-average particle diameters obtained using PSDA (d(PSDA)) compared to TEM (d(TEM)) may be due to the contribution of the double layers and, for MAA-containing particles, swelling by water, both of which would increase the size measured by PSDA (and PCS), but would be absent in the TEM measurements. The hydrodynamic diameter (dh) values are larger than the d(PSDA) and d(TEM) values due to the polydispersity. This is because dh is heavily weighted towards larger particles.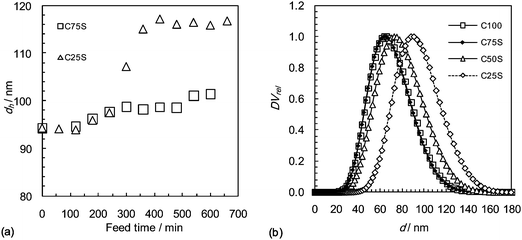 | ||
| Fig. 1 (a) Measured hydrodynamic diameters during growth of the shells for the CxS particles. Data were obtained only at the beginning and end of shell growth for C50S and are not shown. (b) Relative differential volume as a function of diameter for the final CxS and C100 nanoparticles. The data for C100 and C75S overlap. | ||
To establish whether controlled growth of the C100 seed particles had taken place, the total number of particles in the reactor, Np, was calculated for the initial and final dispersions using number-average particle diameters, d(TEM), determined from analysis of TEM micrographs since (for the reasons indicated above) these values more accurately represent the particles than the diameters obtained by PCS and PSDA and because number-average based particle numbers relate more correctly to the kinetics of emulsion polymerisation. Inspection of Table 1 shows that, for each CxS nanoparticle dispersion, the ratio of the final to initial Np value (Np(end)/Np(init)) is reasonably close to unity and that the number-average particle diameter, d(TEM)cal, calculated for growth of the seed particles without secondary nucleation or coagulation is very close to the measured d(TEM) value (certainly within the limits of experimental error). These data provide strong evidence that controlled growth of the seed particles was achieved. Controlled particle growth does not, of course, guarantee control of internal particle morphology. However, given that poly(Bd-co-MAA) is much more hydrophilic than poly(Bd) and that Bd homopolymers and copolymers are known to become partially crosslinked during emulsion polymerization,30 the expectation is that the poly(Bd-co-MAA) will be present in an outer region (‘layer’) of the particles.31,32 As the target shell thickness diminishes, this outer poly(Bd-co-MAA) layer will become more diffuse, but nevertheless, there still will be a distinct outer region where the MAA COOH groups will be concentrated. As will become evident from our studies of these materials, all the experimental data provide evidence that the nanoparticles do indeed have a poly(Bd)–poly(Bd-co-MAA) core–shell structure.
TEM micrographs of the negatively stained particles showed uniform contrast. (Representative TEM micrographs and size distributions are shown in Fig. S1 and S2†, respectively.) Despite using several staining techniques it was not possible to differentiate clearly between the core and shell phases of the nanoparticles, but it was possible to determine the overall particle diameters with accuracy. The relatively high polydispersity for the CxS nanoparticles originates from the C100 seed nanoparticles. Several approaches were used to reduce the size dispersity of the seed particles without success. Nevertheless, the relatively broad size distributions do not prevent structure–property insights being obtained.
The particle size data shown in Table 1 enabled the average layer thicknesses of the shells (δ) to be calculated assuming that the core diameter was constant (Fig. 2). The PCS, PSDA and TEM data all show that the average δ value decreases with increasing ϕc. The δ values determined from the d(TEM)cal values (closed diamonds in Fig. 2) are close to the average of the δ values (not shown) calculated using the dh, d(PSDA), and d(TEM). These δ values (closed diamonds in Fig. 2) are therefore considered to be the most meaningful for the present systems.
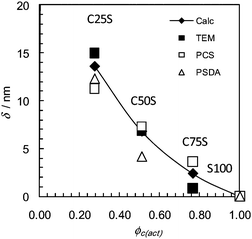 | ||
| Fig. 2 Shell thicknesses obtained using the particle sizes in Table 1. The data are plotted against the actual core volume fractions. The closed diamonds represent calculated δ based on d(TEM)cal values from Table 1. δ was calculated using δ = (dcs − dc)/2, where dcs and dc are the diameters for the core–shell particles and the core, respectively. | ||
Our earlier work involving conventional poly(Bd-co-MAA) nanoparticles showed that the pKa decreased as the MAA concentration increased.26Potentiometric titrations of the dispersions (using aqueous KOH solution) were used here to determine the apparent pKa values for the MAA repeat units in the CxS nanoparticles and indirectly probe the MAA concentration. The apparent pKa was calculated using33,34
 | (1) |
 | ||
| Fig. 3 (a) Variation of pKa with nominal mole fraction of MAA within the nanoparticle shell. (b) Variation of layer thickness determined from PCS measurements with pH for the CxS nanoparticles. The vertical lines are the apparent pKa values from (a). | ||
It can be seen from Fig. 3(a) that the apparent pKa decreases linearly with the logarithm of xMAA(sh). This general effect was reported earlier for conventional poly(Bd-co-MAA) nanoparticles26 and was ascribed to a combination of decreasing hydrophobic attraction between the poly(Bd) segments and increased dissociation of RCOOH groups due to an increasing local dielectric constant. Presumably, a similar explanation applies for the CxS nanoparticles. Hence, the decrease in pKa with xMAA(sh) provides experimental support for a progressively higher MAA content in the shell as ϕc increases.
To further probe the outer layer of the nanoparticles they were tested for pH-triggered swelling using PCS measurements as a function of pH. The dh values for the CxS nanoparticles measured at different pH values (and the non-swollen values) shown in Table 1 were used to estimate the hydrodynamic layer thickness (δh) as a function of pH (see Fig. 3(b)). Generally, swelling occurs when the pH is equal to the respective pKa values. However, for the C50S and C25S nanoparticles there also is evidence of some swelling at pH values significantly below the respective pKa values. This is consistent with the presence of regions of locally high MAA concentrations. The pH-triggered δh increase is greatest for the shells with the highest xMAA(sh), i.e., C75S, which is logical.
DMTA measurements of cast nanoparticle films were conducted to probe the thermally triggered relaxation of molecular motions of the constituent polymer phases.35 The data shown in Fig. 4 gave glass transition temperature (Tg) values from the locations of the tan δ maxima. Two principal Tg values are evident (Table 2) for these non-neutralised CxS nanoparticle films: (i) a Tg value due to the poly(Bd) core phases (Tg(c)), which occurs at −70 °C (labelled A in Fig. 4); and (ii) a higher Tg value that moves to higher temperatures as ϕc increases (labelled B). The latter is attributed to the poly(Bd-co-MAA) shell phases (Tg(sh)). The increase of Tg(sh) with ϕc is ascribed to an increase in the shell MAA concentration.
| Code | T g(c) a/°C | T g(sh) a/°C | x MAA(sh,D) b | x MAA(sh) c |
|---|---|---|---|---|
| a T g(c) and Tg(sh) are the Tgs of the poly(Bd) core and poly(Bd-co-MAA) shell, respectively, determined from the positions of the maxima or shoulders in Fig. 4. b Mole fraction in shell calculated using the Fox equation from the DMTA data. c Nominal values for xMAA in the shell based on the compositions used for preparation (Table 1). | ||||
| C100 | −70 | — | — | — |
| C75S | −69 | −1.0 | 0.30 | 0.165 |
| C50S | −70 | −47 | 0.10 | 0.079 |
| C25S | −70 | −57 | 0.065 | 0.052 |
 | ||
| Fig. 4 Effect of temperature on tan δ for nanoparticle films. To aid comparison and improve clarity the tan δ values for each curve were normalised by dividing each tan δ value by the respective maximum value and then shifted vertically. (A) and (B) show the Tgs for the poly(Bd) core and poly(Bd-co-MAA) shell, respectively. | ||
Assuming that the higher temperature Tg is due to an outer shell of poly(Bd-co-MAA) copolymer it is possible to use the tan δ maxima to estimate the xMAA within this shell (xMAA(sh,D)) for each CxS film. It is implicitly assumed that the film is composed of nanoparticles that retain their structures (core and shell). This is a reasonable assumption because the film formation procedure for the non-neutralised films involved pH values less than 8.0 which meant that the particle shells were in the collapsed state (see Fig. 3(b)). The xMAA(sh,D) values were calculated using the Fox equation,36 the Tg values for the poly(Bd) core (−70 °C) and that reported for poly(MAA) (228 °C, ref. 37). The xMAA(sh,D) values indicate a relatively high MAA content within the high Tg phase (Table 2), which is assumed to originate from the shells, compared to the respective nominal xMAA(sh) values. When these xMAA(sh,D) values are considered together with the pKa and PCS data (Fig. 3) they support the view that the high Tg phase is located in the shells of the nanoparticles within the films.
AFM was used to probe the topography of the nanoparticle films. Phase contrast images are shown in Fig. 5(a)–(c). The phase images represent the phase lag of the output signal with respect to the driving (piezoelectric) signal across the x–y surface. The changes in phase represent differences in the viscoelastic properties (or energy dissipation) of the surface38 with more dissipative phases giving darker images. Distinct nanoparticles can be seen which supports the assumption (above) that the nanoparticle structures retain their integrity upon film formation.
 | ||
| Fig. 5 (a–c) Phase contrast AFM images of nanoparticle film surfaces. Each image has dimensions of 2 × 2 µm. (d) Depiction of the average core–shell particle nanoparticle structures based on the particle size data (see text). The nanoparticle structures are drawn to scale. The core and shell are poly(Bd) and poly(Bd-co-MAA), respectively. | ||
The combination of the core diameter from PSDA (Table 1) and the calculated δ values (Fig. 2) can be used to propose average nanoparticle structures for the CxS nanoparticles used in this study and these are shown in Fig. 5(d). All the evidence points to soft core–shell nanostructures.
3.2. Effects of neutralisation on the morphology of core–shell nanoparticle ionomer films
Neutralisation was achieved using pH-adjusted (pH = 8.0) mixed dispersions of CxS nanoparticles and ZnO particles (Scheme 1). The KOH solution was added to enable ZnO (in the form of a particulate dispersion) to be added to the polymer dispersion without causing colloidal instability. Under these conditions, the ZnO dissolves into the polymer dispersion, forming the Zn2+ salt of the polymerised MAA. This is a new dispersion method to the academic literature for introducing ionic crosslinking and is different to those used to prepare conventional ionomers.35,39,40Representative AFM phase images for CxS films with an α of 112% are shown in Fig. 6(a)–(c). These films have enhanced phase contrast compared to the non-neutralised films (Fig. 5(a)–(c)), i.e., the nanoparticle boundaries are more clearly evident when neutralised. This suggests an increased modulus of the percolating nanoparticle shell phase. Furthermore, the CxS nanoparticle films are not homogeneous on the scale of ca. 100 nm. This is further supported by AFM of freeze-fractured cross-sections of films. A representative height image for a C75S film with an α of 34% is shown in Fig. 6(d). Particle elongation is evident, which is attributed to deformation during the freeze-fracture process, and nanoparticle coalescence is limited. Instead, the film interior consists of well-defined nanoparticles surrounded by an interconnected phase that is consistent with neutralised poly(Bd-co-MAA) from the outer regions of the particles and with percolation of this phase within the film. This is a key morphological difference separating these nanostructured core–shell ionomer films from conventional ionomers.14,19
 | ||
| Fig. 6 Phase contrast AFM images of nanoparticle film surfaces with α = 112% are shown in (a–c). An AFM height image of a freeze-fractured cross-sectional surface for a C75S film with α = 34% is shown in (d). Each image has dimensions of 2 × 2 µm. | ||
DMTA data were obtained for the nanoparticle films as a function of neutralisation. Selected data are shown in Fig. 7. The data for each film as a function of α also were obtained (see Fig. S4†). The plots show the presence of poly(Bd) (labelled as A) and poly(Bd-co-MAA) (labelled as B). Compared to the non-neutralised films (Fig. 4) as α increases a new maximum emerges at temperatures greater than about 0 °C (labelled as C). As α increases the tan δ maximum moves to higher temperatures. Ionic crosslinking increases the Tg for conventional ionomers35 due to increased restrictions on chain mobility. The present trend is attributed to an ionically crosslinked phase in the film that contains Zn2+, i.e., a poly(Bd-co-MAA)/Zn2+.
 | ||
| Fig. 7 Effect of temperature on tan δ for nanoparticle films which have α of (a) 53% and (b) 112%. The tan δ values for each curve were normalised and shifted vertically. (A) to (C) represent Tgs for the poly(Bd) core, poly(Bd-co-MAA) shell and outer poly(Bd-co-MAA)/Zn2+ shell of the nanoparticles that comprise the film, respectively. | ||
The DMTA data shown in Fig. 7 (and Fig. S4†) provide clear evidence for three different glass transitions and hence three distinct phases within the films cast from the CxS nanoparticles. For films cast from the C25S and C50S nanoparticles, this implies that poly(Bd), poly(Bd-co-MAA) and poly(Bd-co-MAA)/Zn2+ are present. For C75S only two phases are discerned: poly(Bd) and poly(Bd-co-MAA)/Zn2+ for all α values studied. We can be confident that the shells of the nanoparticles were collapsed during film formation because a pH of 8.0 is not conducive to swelling of the shell (Fig. 3(b)). Consequently, ingress of Zn2+ would have been restricted due to the collapsed shell. Given that the shells of the nanoparticles are MAA-rich it follows that the poly(Bd-co-MAA)/Zn2+ phase resides within the outer shells of the nanoparticles.
Whilst one has to be cautious in relating tan δ peak heights with proportions of each phase of a multiphase system, it is possible to obtain qualitative information that can be instructive.35 Inspection of the data shown in Fig. 7 reveals that the proportion of non-neutralised poly(Bd-co-MAA) phase (B) decreases and the proportion of poly(Bd-co-MAA/Zn2+) phase (C) increases with α. This also can be seen from the full dataset (Fig. S4†) and may indicate that the thickness of poly(Bd-co-MAA)/Zn2+ outer shell increases with α. Importantly, even when the nominal degree of neutralisation (α) was 112% (Fig. 7(b)) there is significant non-neutralised poly(Bd-co-MAA) phase present. For conventional ionomers neutralisation occurs more uniformly (e.g., all components in solution) and can result in a change of the majority phase from being non-ionically crosslinked to crosslinked (phase inversion).35 This cannot occur in the present core–shell nanoparticle systems and is another factor that distinguishes them from conventional ionomers.35
In order to probe the global changes in neutralisation ATR FTIR spectroscopy of the films was used. This technique has been applied to conventional ionomers containing RCOOH groups.41,42Fig. 8(a) shows representative spectra for C50S films. Bands were present at 1700, 1640 and 1600–1550 cm−1. They are due to, respectively, hydrogen bonded acid pairs,41 –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–42 and Zn2+ (and K+) acid salts. The latter cations are ionically bonded to RCOO−. The band at 1585 cm−1 is due to41 tetrahedrally coordinated Zn2+. It is evident that as α increases the intensity of the RCOOH band (1700 cm−1) decreases whilst that for the ionic species (1550–1600 cm−1) increases.
CH–42 and Zn2+ (and K+) acid salts. The latter cations are ionically bonded to RCOO−. The band at 1585 cm−1 is due to41 tetrahedrally coordinated Zn2+. It is evident that as α increases the intensity of the RCOOH band (1700 cm−1) decreases whilst that for the ionic species (1550–1600 cm−1) increases.
 | ||
| Fig. 8 (a) FTIR spectra for C50S films showing the intermediate wavenumber regions. Only representative spectra have been shown to aid resolution of the main features. (b) Variation of αFTIR values with the nominal α values for the S100, C75S, C50S and C25S films. The dotted line in (b) shows when αFTIR is equal to α. | ||
The FTIR spectra were used to estimate the overall extents of neutralisation (αFTIR). The area ratio for the RCOOH groups and –CHCH– groups, i.e., ACOOH/ACC, was plotted against α (Fig. S5†) and then normalised. The following equation was used to estimate neutralisation from the FTIR data:
 | (2) |
C values were normalised using the value for ACOOH/ACC obtained at 0% neutralisation. The results of this analysis are plotted in Fig. 8(b). Interestingly, αFTIR is less than the expected value at high α values, especially for the films prepared using nanoparticles with the thickest shells (C25S and S100). This is consistent with the trends established above from the DMTA data and confirms that incomplete neutralisation of poly(Bd-co-MAA) occurred with those particles.
The variation of Tg with neutralisation provides useful information about the useable temperature range for the films. The variation of Tg for the poly(Bd-co-MAA)/Zn2+ shell as a function of αFTIR appears in Fig. 9. The increase in Tg with αFTIR is very similar for C50S and C25S. Interestingly, the Tg values for all three film types coincide at about αFTIR = 50%. Presumably, at that degree of neutralisation all of the poly(Bd-co-MAA+)/Zn2 phases have similar ionic crosslink densities. It is noted that the film prepared using nanoparticles with the thinnest shells (C75S) has a much more pronounced increase in Tg beyond 50% neutralisation.
 | ||
| Fig. 9 Glass transition temperature of the outer poly(Bd-co-MAA)/Zn2+ shell as a function of the neutralisation determined by FTIR for CxS nanoparticle films. | ||
The average rate of Tg increase for the films composed of thick-shell nanoparticles (C25S and C50S) (Fig. 9) is about 1 °C per mol% ion content. This is a much lower value than reported for conventional ionomers. For example Ma et al.35 reported 5.5 °C per mol% ion content for PMMA/MAA ionomers. This indicates that the Zn2+ ions are less effective at restricting chain mobility in these films and is an indication of differences in the structures of the crosslinking species (multiplets cf.clusters).19 A more effective ionic crosslinking is indicated for the C75S film (thinner shell) at αFTIR values greater than 50% by the enhanced abrupt increase in the gradient in that region.
Using all of the data presented in this section we can propose a general structure for the C25S and C50S nanoparticles with an α value of 112% (see Fig. 10). For the C25S nanoparticles the poly(Bd-co-MAA) shell would be thickest. The C75S particles would only have a poly(Bd) core and a poly(Bd-co-MAA/Zn2+) shell. Because shell swelling did not occur under the conditions used for film casting the extent of nanoparticle coalescence is limited, as indicated from AFM (Fig. 6).
 | ||
| Fig. 10 Depiction of the proposed structure of the core–shell C25S and C50S nanoparticles (α = 112%) and also the proposed honeycomb morphology of the cast films. | ||
3.3. Relationship between the mechanical properties of nanostructured ionomers and nanoparticle shell composition
In this section we explore the relationships between nanoparticle structural variation and mechanical properties of the cast films. The cast films are robust when dry and do not redisperse when placed in water. The storage modulus (E′) was measured at 25 °C using DMTA over a range of α values and the data appear in Fig. 11(a). The value for E′ increases at a given αFTIR value with increasing ϕc. The increased MAA contents within the shells (xMAA(sh.D)) of these particles (Table 2) is responsible for the increase in E′. It is interesting to compare the data for C75S and S100 because the nanoparticles had the same overall MAA mole fraction on a whole particle basis (0.04) and yet have very different modulus values. An E′ increase of up to a factor of eight can be achieved by confining the MAA to a boundary with a thin outer poly(Bd-co-MAA) shell. This demonstrates the potential to tune the mechanical properties of the films using core–shell nanoparticle architecture. This is similar to recent work involving covalent crosslinking of shell phases in soft core–shell nanoparticles during film formation.43 However, in the present work we used ionic crosslinking which has considerable versatility in terms of the conditions in which it can be employed. | ||
| Fig. 11 Effect of neutralisation on (a) the storage modulus and (b) E′/xMAA(sh,D) at 25 °C. The legend is the same for both graphs. | ||
The E′ values for the films generally increased with the Tg of the outer poly(Bd-co-MAA)/Zn2+ shell (see Fig. S6†) implying that ionic crosslinking due to the Zn2+ at the periphery of the nanoparticles was responsible. According to rubber elasticity theory the elastic modulus should be proportional to the number density of elastically effective chains44 (n). In the present case the crosslinks that produce elastically effective chains involve ionically crosslinked MAA groups (RCOO−) which almost certainly are located in the outer layer of the nanoparticles. If this is correct (and assuming that the elasticity of the films is dominated by the crosslinked shells) the value for n should be proportional to xMAA(sh). Accordingly, E′/xMAA for each film should be similar for each system. This was tested (see Fig. 11(b)). With the exception of the non-neutralised films (αFTIR = 0) the data are similar, although not identical, for each αFTIR value. Furthermore, for αFTIR values greater than 34%, E′ appears to be proportional to αFTIR. This is different to conventional ionomers where the modulus usually shows a supra-linear dependence on neutralisation23,35 due to cluster formation. The linear behaviour observed here suggests independent multifunctional crosslinks23 are present and not clusters. The presence of these species could also account for the low rate of Tg increase noted above (Fig. 9).
Selected tensile stress–strain properties are shown in Fig. 12(a)–(c). (The complete dataset appears in Fig. S7†.) For non-neutralised films (Fig. 12(a)) elastic behaviour is evident at low strains (λ) followed by plastic deformation at higher strains.
 | ||
| Fig. 12 Tensile data for nanoparticle films measured at 25 °C. Stress as a function of strain for nanoparticle films are shown for nominal α values of (a) 0%, (b) 53% and (c) 112%. Note that for (c) the C75S films were not of sufficient quality to enable investigation by this method. Note the change of y-axis of (a) compared to (b) and (c). The tensile modulus values divided by xMAA(sh,D) as a function of αFTIR are shown in (d). | ||
The data in Fig. 12(a)–(c) show that neutralisation dramatically changes the σvs.λ behaviours and that the σ values increase strongly with α. Neutralisation introduces pronounced strain hardening for the C25S and S100 films at high λ values. Once again our new ionically crosslinked systems give data that are consistent with previous observations on films formed from soft core–shell nanoparticles with covalently crosslinked shell phases43 and also with theoretical expectation.32
The tensile modulus values at 25 °C were tested for their dependence on xMAA(sh) by plotting (E25(Tens)/xMAA(sh,D)) against αFTIR (Fig. 12(d)). Similar general trends to those shown in Fig. 11(b) are apparent. A linear regime for these data is evident between αFTIR of 25 and 100%, i.e., E25(Tens) appears to be proportional to αFTIR. These data confirm that shell crosslinking plays a major role in controlling the mechanical properties of these new nanostructured ionomers and that the elastic modulus values are tuneable using xMAA or α.
Conventional synthetic butadiene rubber films are well known to exhibit hardening on aging45,46via a free-radical mechanism and the antioxidant Wingstay L is often added to improve stability. Wingstay L is the reaction product of butylated p-cresol and di-cyclo pentadiene.45 Because aging is important for many industrial applications the stability of the films to heating at 100 °C for 22 h was investigated. The stress–strain data for as-made films and also films containing the Wingstay L are shown in Fig. S8†. Addition of the antioxidant removed the hardening effect and gave films with good thermal stability. This is consistent with work reported for conventional rubber films.45
Finally, we compare the mechanical properties of our nanostructured ionomers to conventional ionomers. The closest conventional ionomer system that we could find was the Krynac 7.40/Krynac 2750 system reported by Ibarra and Alzorriz.47 which contained 27% acrylonitrile (AN). Krynac 7.4 contained 7 wt% of –COOH groups48 and was diluted with47Krynac 2750 to achieve a range of –COOH contents. Zn2+ was added, in the form of ZnO2/ZnO, by solid state mixing. In their study the amount of Zn2+ was sufficient to neutralise all of the –COOH groups. Fig. 13 shows the variation of the stress at λ = 1.0 (σ1.0) as a function of the mole fraction of –COOH groups (xCOOH) for samples from our study and theirs.47 These data highlight the fact that for our nanostructured ionomers a major increase in σ1.0 can be achieved at constant xCOOH. Clearly, that is not possible for conventional ionomers. This comparison demonstrates the unique benefit that can be achieved through nanostructuring.
 | ||
| Fig. 13 Comparison of stress at λ = 1.0 for nanostructured (closed diamonds) and conventional (open triangles) ionomers as a function of the mole fraction of COOH groups (xCOOH) present. The conventional ionomer xCOOH was calculated from the weight fractions of –COOH (WCOOH) reported in ref. 47 using, xCOOH = (2.22WCOOH)/(1.85 + 0.37WCOOH). The data for the nanostructured ionomers were taken from the stress–strain data shown in Fig. 12(c). | ||
4. Conclusions
The structure–property relationships of new soft core–shell poly(Bd)/poly(Bd-co-MAA) nanoparticles and films cast from them have been investigated. The cast films in which –COOH groups are neutralised with Zn2+ represent a new class of ionomers which are nanostructured and with mechanical properties that depend strongly on the MAA content and degree of neutralisation. At first glance this might seem similar to conventional ionomers.14,18,19,23 However, for these nanostructured ionomers it is the structure of the shells of the core–shell nanoparticles that controls the modulus values. (Conventional ionomers have modulus values controlled by the bulk phase.) The ionomer films studied here have different nanometre-scale structures to conventional ionomers. The core–shell nanoparticle building blocks in the film force heterogeneity on the nanometre scale of the shell regions. Our data point to a honeycomb type structure that is Zn2+-rich and percolates the films (Fig. 10). The ionic crosslinks are located within this continuous phase and contribute to both the intra- and inter-particle crosslinking. In this phase the Zn2+ ions are tetrahedrally coordinated which is similar to the bonding reported in conventional ionomers. However, for the films prepared from thicker shell nanoparticles (C25S and C50S) the crosslinking is less efficient than in conventional ionomers and this leads to a reduced rate of change of Tg with mol% neutralisation. Interestingly, there are significant proportions of non-neutralised MAA groups present which are most likely buried beneath the external poly(MMA-co-MAA)/Zn2+ shells and is probably a consequence of the dispersion preparation method. A multi-shell structure was suggested for these nanoparticles within the cast films. Like conventional ionomers the mechanical properties of these nanostructured ionomers are tuneable. However, in contrast to conventional ionomers, the modulus values are generally proportional to the degree of neutralisation and also the mole fraction of MAA in the nanoparticle shells. This implies tuneability through the design of core–shell architecture of the nanoparticles, which is a new concept for ionomers.The core–shell nanoparticle structures also produced major changes in film properties when compared to films prepared using conventional nanoparticles prepared by monomer-flooded batch emulsion copolymerisation (S100). The introduction of a thin boundary (for C75S) resulted in the greatest modulus increase. The modulus values for C75S increased by up to a factor of eight compared to S100. In this regard films prepared using the core–shell nanoparticles are superior to those prepared using conventional nanoparticles. The colloidal approach used here enables cast films with tuneable mechanical properties to be prepared and should be generally applicable to any soft core–shell nanoparticle dispersion (or conventional latex) where the outer shell (or outer surface) of the nanoparticles contains COOH groups. Since the dispersions are water based and, in principle, a range of divalent cations could be used19 this general approach should have potential application in surface coatings, pressure sensitive adhesives and biomaterials research.
Acknowledgements
The authors gratefully acknowledge The Royal Thai Government and Synthomer Ltd., UK for funding this work. We thank Dave Hui for technical assistance.References
- I. Berndt and W. Richtering, Macromolecules, 2003, 36, 8780 CrossRef CAS.
- F. Deplace, M. A. Rabjohns, T. Yamaguchi, A. B. Foster, C. Carelli, C.-H. Lei, K. Ouzineb, J. L. Keddie, P. A. Lovell and C. Creton, Soft Matter, 2009, 5, 1440 RSC.
- N. Dingenouts, C. Norhausen and M. Ballauff, Macromolecules, 1998, 31, 8912 CrossRef CAS.
- M. Okubo, Y. Lu and Z. Wang, Colloid Polym. Sci., 1999, 277, 77 CrossRef CAS.
- G. Petekidis, J. Gapinski, P. Seymour, J. S. van Dujineveldt, D. Vlassopoulos and G. Fytas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 69, 042401 CrossRef CAS.
- E. van Ruymbeke, A. Pamvouxoglou, D. Vlassopoulos, G. Petekidis, G. Mountrichas and S. Pispas, Soft Matter, 2010, 6, 881 RSC.
- S. A. F. Bon, S. Cauvin and P. J. Colver, Soft Matter, 2007, 3, 194 RSC.
- P. Chambon, A. Chemtob, E. Cloutet, H. Cramail, S. Gibanel, Y. Gnanou, V. Heroguez, D. Quemener and B. Radhakrishnan, Polym. Int., 2006, 55, 1146 CrossRef CAS.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1 CrossRef CAS.
- P. A. Lovell, in Emulsion Polymerisation and Emulsion Polymers, ed. P. A. Lovell and M. El-Aasser, John Wiley and Sons, Chichester, 1997, ch. 7 Search PubMed.
- B. E. Rodriguez, M. S. Wolfe and M. Fryd, Macromolecules, 1994, 27, 6642–6647 CrossRef CAS.
- L. Goehring, W. J. Clegg and A. F. Routh, Langmuir, 2010, 26, 9269 CrossRef CAS.
- H. N. Yow, I. Beristain, M. Goikoetxea, M. J. Barandiaran and A. F. Routh, Langmuir, 2010, 26, 6335 CrossRef CAS.
- A. Eisenberg, Macromolecules, 1970, 3, 147–154 CrossRef CAS.
- M. R. Moghbell, N. Mohammadi and R. Bagheri, J. Appl. Polym. Sci., 2007, 105, 1412 CrossRef.
- M. Toshihiko, Appl. Surf. Sci., 2006, 252, 7018 CrossRef.
- J. E. Kennedy, J. G. Lyons, L. M. Geever and C. L. Higginbotham, Mater. Sci. Eng., C, 2009, 29, 1655 CrossRef CAS.
- I. Capek, Adv. Colloid Interface Sci., 2004, 112, 1 CrossRef CAS.
- A. Eisenberg and J.-S. Kim, Introduction to Ionomers, John Wiley & Sons, New York, 1998 Search PubMed.
- J.-S. Kim, G. Wu and A. Eisenberg, Macromolecules, 1994, 27, 814 CrossRef CAS.
- C. T. Meyer and M. Pineri, Polymer, 1976, 17, 382 CrossRef CAS.
- M. Pineri and C. Meyer, J. Polym. Sci., Polym. Phys. Ed., 1974, 12, 115 CrossRef CAS.
- I. Capek, Adv. Colloid Interface Sci., 2005, 118, 73 CrossRef CAS.
- W. Wang, W. Liu, G. J. Tudryn, R. H. Colby and K. I. Winey, Macromolecules, 2010, 43, 4223 CrossRef CAS.
- O. Pinprayoon, A. Saiani, R. Groves and B. R. Saunders, J. Colloid Interface Sci., 2009, 336, 73 CrossRef CAS.
- O. Pinprayoon, R. Groves and B. R. Saunders, J. Colloid Interface Sci., 2008, 321, 315 CrossRef CAS.
- S. R. Turner, R. A. Weiss and R. D. Lundberg, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 535 CrossRef CAS.
- R. A. Weiss, R. D. Lundberg and S. R. Turner, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 549 CrossRef CAS.
- R. A. Weiss, S. R. Turner and R. D. Lundberg, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 525 CrossRef CAS.
- D. C. Blackley, in Emulsion Polymerisation and Emulsion Polymers, ed. P. A. Lovell and M. S. El-Asser, John Wiley & Sons, 1997, ch. 15 Search PubMed.
- J. M. Stubbs and D. C. Sundberg, Prog. Org. Coat., 2008, 61, 156 CrossRef CAS.
- V. L. Dimonie, E. S. Daniels, O. L. Shaffer and M. S. El-Asser, in Emulsion Polymerisation and Emulsion Polymers, ed. P. A. Lovell and M. S. El-Asser, John Wiley & Sons, 1997, ch. 9 Search PubMed.
- O. E. Philippova, D. Hourdet, R. Audebert and A. R. Khokhlov, Macromolecules, 1997, 30, 8278 CrossRef CAS.
- R. D. Porasso, J. C. Benegas and M. A. G. T. van den Hoop, J. Phys. Chem. B, 1999, 103, 2361 CrossRef CAS.
- X. Ma, J. A. Sauer and M. Hara, Macromolecules, 1995, 28, 3953 CrossRef CAS.
- K. L. Mittal, Polyimides and Other High Temperature Polymers—Synthesis, Characterisation and Applications, VSP, 2007, vol. 4 Search PubMed.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley & Son, New York, 4th edn, 1999 Search PubMed.
- B. Anczykowski, B. Gotsmann, H. Fuchs, J. P. Cleveland and V. B. Elings, Appl. Surf. Sci., 1999, 140, 376 CrossRef CAS.
- K. Han and H. L. Williams, J. Appl. Polym. Sci., 1991, 42, 1845 CrossRef CAS.
- C. W. A. Ng and W. J. MacNight, Macromolecules, 1996, 29, 2421 CrossRef CAS.
- B. A. Brozoski, P. C. Painter and M. M. Coleman, Macromolecules, 1984, 8, 1591 CrossRef.
- U. K. Mandal, Polym. Int., 2000, 49, 1653 CrossRef CAS.
- A. B. Foster, P. A. Lovell and M. A. Rabjohns, Polymer, 2009, 50, 1654 CrossRef CAS.
- W. Schlesing, M. Buhk and M. Osterhold, Prog. Org. Coat., 2004, 49, 197 CrossRef CAS.
- N. Martakis and M. Niaounakis, J. Appl. Polym. Sci., 1992, 46, 1737 CrossRef CAS.
- J. R. Shelton, F. J. Wherley and W. L. Cox, Ind. Eng. Chem., 1954, 45, 2080.
- L. Ibarra and M. Alzorriz, J. Appl. Polym. Sci., 2002, 84, 606.
- L. Ibarra, A. Rodriquez and I. Mora, Eur. Polym. J., 2007, 43, 753 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI contains transmission electron micrographs of the nanoparticles (Fig. S1), their particle size distributions (Fig. S2), a graph showing pKa vs. degree of neutralisation (Fig. S3), DMTA data for films with different neutralisations (Fig. S4), Variation of area ratio from FTIR with neutralisation (Fig. S5), Variation of elastic modulus with glass transition of the films (Fig. S6), stress vs. strain for the films at different neutralisations (Fig. S7) and Stress vs. strain for heat-treated films (Fig. S8). See DOI: 10.1039/c0sm00447b |
| This journal is © The Royal Society of Chemistry 2011 |