AOT vesicles as templates for the horseradish peroxidase-triggered polymerization of aniline
Zengwei
Guo
a,
Nicole
Hauser
a,
Aitor
Moreno
b,
Takashi
Ishikawa
c and
Peter
Walde
*a
aDepartment of Materials, ETH Zürich, Wolfgang-Pauli-Str. 10, CH-8093, Zürich, Switzerland. E-mail: peter.walde@mat.ethz.ch; Fax: +41 44 63 21265; Tel: +41 44 63 20473
bDepartment of Chemistry and Applied Biosciences, Wolfgang-Pauli-Str. 10, CH-8093, Zürich, Switzerland
cDepartment of Biology, ETH Zürich, Schafmattstrasse 20, CH-8093, Zürich, Switzerland
First published on 5th November 2010
Abstract
In dilute aqueous solution at pH = 4.3 in the presence of 0.1 M sodium dihydrogen phosphate, AOT (bis-(2-ethylhexyl)sulfosuccinate) was found to form vesicles. The average diameter of the vesicles was adjusted to about 70 nm by polycarbonate membrane extrusion. The vesicles were applied as chemical structure-controlling templates for the horseradish peroxidase/H2O2-triggered polymerization of aniline to yield the green emeraldine salt form of polyaniline. The enzyme-containing vesicular reaction system was optimized with respect to obtaining a reaction product with high absorbance in the NIR region of the spectrum which is known to be a characteristic property of the conductive emeraldine salt form of polyaniline. The reaction system was analyzed by cryo transmission electron microscopy, 1H NMR, UV/VIS/NIR, circular dichroism and fluorescence measurements. The peroxidase was found to be bound to the vesicles leading to an initiation of the reaction preferentially on the vesicles surface and not in the bulk aqueous solution. Before the reaction was started by H2O2 addition, the anilinium cations were found to only weakly interact with the surface of the vesicles. After polymerization, a stable suspension containing vesicles which were coated with polyaniline was obtained. The reaction product was isolated and analyzed by FTIR measurements. With respect to the vesicle system used previously, SDBS/decanoic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Z. Guo, H. Rüegger, R. Kissner, T. Ishikawa, M. Willeke and P. Walde, Langmuir, 2009, 25, 11390–11405), the AOT system has several advantages for further explorations of this type of in situ formation of conductive vesicle-based polymer capsules.
:1) (Z. Guo, H. Rüegger, R. Kissner, T. Ishikawa, M. Willeke and P. Walde, Langmuir, 2009, 25, 11390–11405), the AOT system has several advantages for further explorations of this type of in situ formation of conductive vesicle-based polymer capsules.
1. Introdution
The oxidative polymerization of aniline can lead to a number of different polymeric products which may not only differ in the degree of oxidation and protonation but also in the level of branching, on the nature of the chemical bond between the aniline monomers and on the morphology of the polymerization products obtained.1 With respect to the polymerization conditions, the chemical structure of the polyaniline (PANI) obtained, its morphology and physico-chemical properties depend on many factors. These include the type of oxidant used, the concentrations of aniline and oxidant and their molar ratio, the temperature and acidity.2 If H2O2 and the enzyme horseradish peroxidase (HRP) are used as oxidants, the green electrically conductive emeraldine salt form of PANI can easily be obtained at room temperature under mild, environmentally friendly conditions in slightly acidic aqueous medium (pH = 4.3).3 One of the key requirements is, however, that an appropriate chemical structure-controlling template4 is added, as originally demonstrated by Samuelson and coworkers.5The template can be a negatively charged polymer (particularly poly(styrene sulfonate)),5–7 negatively charged micelles,6,8 negatively charged vesicles,9 or even negatively charged inorganic particles.10 It seems that the template plays several roles. The template may concentrate and pre-orient the positively charged anilinium ions, possibly via N–H⋯O–S hydrogen bonds, so that the polymerization reaction is regioselective in the sense that the monomers are predominantly added at the para-position with respect to the amino group of aniline. This regioselective coupling determines the amount of linear para-directed units formed as compared to ortho-directed units. Other roles of the template are as efficient counter ion (dopant), most likely again via N–H⋯O–S hydrogen bonds,11 and as complexing agent for the polymerization products to increase the dispersability of the otherwise insoluble PANI.
If the reaction conditions are not optimized, the enzymatic polymerization product obtained is not the desired emeraldine salt (Scheme 1), as can be determined conveniently by UV/VIS/NIR spectroscopy.12,13 Absorption bands in the NIR region of the spectrum (at λ > 750 nm) are typical for the conductive emeraldine salt form of PANI,12–17 while deprotonation (to form the emeraldine base) or overoxidation (to obtain the pernigraniline form) leads to a blue shift with absorption maxima at ∼650 nm (for the emeraldine base)18 and ∼750 nm (pernigraniline salt).9 In the case of the emeraldine salt form of PANI (Scheme 1), the absorption maximum in the NIR region seems to strongly depend on the chain conformation which influences the conjugation length.19 “Compact coils” lead to a localized absorption near 750 nm,19,20 while “expanded coils” show broad absorption bands with maximum absorption centered at around 1000 nm or at even higher wavelengths.22 The fully reduced form of PANI (leucoemeraldine) has absorption maxima below 400 nm.10,12 Furthermore, branched ortho-directed PANI has strong absorption around 550 nm with negligible absorption in the region of 1000 nm.13 Based on this strong chemical structure and conformation dependence of the VIS/NIR spectrum of PANI, absorption measurements are often used as convenient and simple tools for optimizing aniline polymerization conditions. Unfortunately, the low solubility of the emeraldine salt form of PANI makes the determination of the molar mass by size exclusion chromatography and determination of the chemical structure by solution NMR difficult. Furthermore, the (paramagnetic) unpaired electrons expected to be present in the polaron state of the conductive emeraldine salt form of PANI (Scheme 1) hinder NMR measurements even if oligomeric products would be soluble. Often, infrared absorption measurements are used for a confirmation of the presence of the expected structural units in the PANI synthesized (for example benzenoid units show a characteristic absorption at about 1500 cm−1 and quinoid units at about 1590 cm−1 in the emeraldine base form),13,23 although the assignment of all peaks is not always possible unequivocally.24
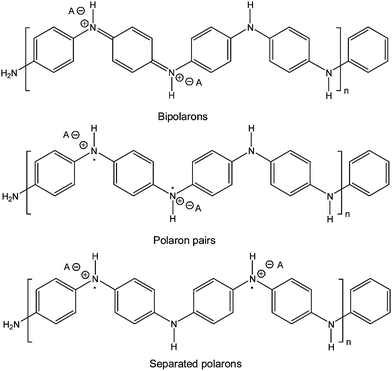 | ||
| Scheme 1 Emeraldine salt form of polyaniline with its bipolaron and polaron states.1f | ||
Recently, we have studied the HRP/H2O2-catalyzed polymerization of aniline in the presence of SDBS/decanoic acid (1:1, mole ratio) vesicles,9 and we found that this type of vesicles is a promising surfactant template since the enzyme was more stable as compared to the enzyme stability in the presence of corresponding micellar templates that were used previously.6,8a Furthermore, we proposed that the localization of the enzyme on the surface of the vesicular templates may be essential for obtaining preferentially the para-linked, linear emeraldine salt form of PANI at pH = 4.3 from the very beginning of the reaction.9 In addition, we carried out NOESY NMR measurements of the vesicle system before polymerization since we were interested in gaining information about the possible association of aniline with the vesicle template before the reaction was initiated. Drawbacks of using SDBS, however, were (i) the presence of an aromatic ring in this surfactant which gave broad aromatic signals that overlapped with the signals of the aromatic aniline protons and (ii) the fact that commercial SDBS is actually a mixture of surfactants.9 Furthermore, below a temperature of about 10 °C, SDBS/decanoic acid (1:1) vesicle precipitation was observed since the phase transition temperature of this surfactant mixture was found to be at 9 °C.9 This was another clear drawback which hindered carrying out HRP-catalyzed polymerization reactions in the presence of the vesicles at low temperature. Template-assisted polymerization reactions at low temperature may be advantageous since they may lead to more uniform polymerization products due to a decreased aniline mobility on the vesicle surface as compared to 25 °C, i.e. a more ordered aniline pre-orientation before polymerization is started.
In this work we have used vesicles from AOT, sodium bis-(2-ethylhexyl)sulfosuccinate (Scheme 2), which do not have the mentioned drawbacks of the SDBS/decanoic acid (1:1) system, i.e. (i) no aromatic ring in the chemical structure, (ii) no chemical heterogeneity in the surfactant sample apart from the presence of different stereoisomers, and (iii) no precipitation of the vesicles at a temperature between 5 and 10 °C. This latter property allowed studying the HRP/H2O2-catalyzed polymerization reaction under various experimental conditions also below 10 °C.
 | ||
| Scheme 2 Chemical structure of AOT (sodium bis-(2-ethylhexyl)sulfosuccinate). | ||
To the best of our knowledge, this is only the second time that vesicles are used as chemical structure-controlling templates4 for directing an enzymatic polymerization reaction. We hope that the promising data presented in this work stimulate further investigations by others on related systems. With respect to our previous study with SDBS/decanoic acid (1:1) vesicles,9 the current investigation has shown that the AOT vesicle system is clearly superior, as outlined in the following.
2. Materials and methods
2.1. Materials
Horseradish peroxidase (HRP, type II, RZ = 2.0), sodium bis(2-ethylhexyl) sulfosuccinate (AOT, 99.0%), 2-(p-toluidino)-6-naphthalenesulfonic acid potassium salt (TNS) and sodium phosphate (NaH2PO4, ≥99%) were purchased from Sigma. Based on the absorption at 403 nm, the HRP content in the solid product was found to be 54 wt%, taking into account the molar absorbance of HRP, ε403 = 1.02 × 105 M−1 cm−1.25 All HRP concentrations given are in amount of solid product per volume, except for the circular dichroism (CD) measurements (see below). Hydrogen peroxide (30%) was from Merck and aniline (≥99.5%) from Fluka. Horseradish peroxidase isoenzyme C (HRPC, grade I-C, RZ ≥ 3.0) for CD measurements was purchased from Toyobo Enzymes. The HRPC concentration was determined by using the molar absorbance given above.2.2. Preparation of AOT vesicles
AOT vesicles were prepared either by a combined bath/tip sonication procedure or by the freezing–thawing extrusion method with a pH = 4.3 solution containing 0.1 M H2PO4−, as described previously.9 In both cases, a thin film of AOT was first formed by dissolving 0.178 g (0.4 mmol) AOT in 5 mL chloroform in a 50 mL round-bottom flask, followed by chloroform removal with a rotary evaporator under reduced pressure. The film was dried overnight under high vacuum. In the case of sonication, bath sonication (with a Bandelin Sonorex RK 100 H) was carried out for 4 min at room temperature, followed by tip sonication (with a Branson Sonifier 250) for another 4 min at room temperature. In the case of the extrusion method,26 final extrusions were through 100 nm Nucleopore polycarbonate membranes.2.3. Polymerization of aniline by HRP/H2O2 in the presence of AOT vesicles
The polymerization of aniline initiated by HRP/H2O2 in the presence of AOT vesicles was carried out at pH = 4.3 (0.1 M H2PO4−) at room temperature. A volume of 6 mL AOT suspension (20 mM AOT) and 4 mL 40 mM aniline (prepared in 0.1 M H2PO4− solution, pH = 4.3) was added into a 200 mL flask containing a magnetic stirrer bar, together with 108.8 mL 0.1 M H2PO4− solution (pH = 4.3). To this mixture, 0.6 mL of a solution containing 5 mg mL−1 HRP in water was then added. The reaction was finally started by quick addition of 0.6 mL 0.2 M H2O2 (prepared in H2PO4− solution, pH = 4.3). The total reaction volume was 120 mL and the total reaction time 5 h. [AOT] = 1 mM, [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, and [H2O2] = 1 mM. For reactions carried out in quartz cuvettes inside a spectrophotometer, the volumes were reduced accordingly.2.4. UV/VIS/NIR measurements
Absorption measurements in the ultraviolet (UV), visible (VIS) and near infrared (NIR) region of the spectrum were recorded with a Perkin Elmer Lambda 20 or a JASCO V-670 instrument at 25 °C, using quartz cuvettes with a path length of either 0.1 cm or 0.5 cm (from Hellma). UV/VIS measurements of small volume samples (below 0.1 mL) were also measured with a NanoDrop ND1000 instrument from Thermo Scientific.2.5. Circular dichroism (CD) measurements
Circular dichroism (CD) measurements were performed on a JASCO J-715 spectropolarimeter at 25 °C by using HRPC at a concentration of 12 µM. For the CD spectrum recorded in the Soret band region of the spectrum, quartz cuvettes with a path length of 0.5 cm (from Hellma) were used. For the far UV region, the quartz cuvettes had a path length of 0.1 cm. The AOT concentration was 1 mM. The spectra were recorded with a wavelength step of 1 nm and a time constant of 1 s. The reported mean residue ellipticity [θ] is defined as follows: [θ] = θ/(10cln); θ measured ellipticity (in mdeg); c molar concentration of HRPC (in M); l path length of the cuvette (in cm); n: number of amino acids per HRPC (n = 308).272.6. Fluorescence measurements
Fluorescence spectra of TNS as fluorescent membrane probe were recorded on a SPEX Fluorolog 2 instrument from Jobin Yvon (UK) using 1 cm quartz cells, as described previously.9 The excitation and emission slits were set to 0.5 mm and 1 mm, respectively. TNS was dissolved in methanol at a concentration of 3 mM and a small volume of this solution was added to preformed AOT vesicles prepared at pH = 4.3 (0.1 M H2PO4−). The vesicle suspension was stored for 30 min at room temperature to ensure complete TNS incorporation into the vesicle membrane. A solution of aniline or HRP was added to the vesicle suspension, followed by storage in the dark for 1 h at room temperature, before the fluorescence measurements were started. The concentration of TNS was 1 µM in the measurements with aniline and 3.2 µM in the measurements with HRP, [AOT] = 1 mM.2.7. NMR measurements
For the NMR measurements, a phosphate solution in deuterated water was prepared by first dissolving sodium phosphate (Na3PO4) in D2O, followed by adjustment of the pH to 4.3 (pH meter reading, corresponding to pD = 3.9)9 by using 1 M DCl and 1 M NaOD solution. The NMR measurements were carried out on a Bruker Avance 700 MHz instrument. For NOESY measurements, the aniline and AOT concentrations were 8 mM each. For the 1H NMR measurements of the reaction system, the concentrations of AOT and aniline were 1 mM and 1.33 mM respectively; [HRP] = 25 µg mL−1 and [H2O2] = 1 mM.2.8. Cryo-TEM measurements
The cryo transmission electron microscopy (Cryo-TEM) analysis of the AOT vesicles was performed by using vesicle suspensions prepared by the freezing–thawing extrusion method. Details of the sample preparation and of the electron microscopy analysis are described elsewhere.9,28 For the analysis of the AOT vesicles in 0.1 M H2PO4− (pH = 4.3, no reaction), the AOT concentration was 6 mM. For the analysis of the reaction mixture, the following conditions were used: [AOT] = 3 mM, [aniline] = 4 mM, [HRP] = 75 µg mL−1, [H2O2] = 3 mM, T = 7 °C, and reaction time: 12 h. Before analysis by cryo-TEM, the vesicle suspension was concentrated by using a Millipore ultrafiltration unit with a molar mass cut-off of 10000 D and a Hermle Z 320K table centrifuge (30 min, 2000 rpm, 25 °C). The suspension which did not pass through the ultrafiltration membrane (retentate) was analyzed.
2.9. Determination of the amount of remaining aniline in the reaction system
To analyze the amount of aniline which was not converted into PANI during the polymerization reaction, the reaction was carried out with a volume of 20 mL. During the reaction, 0.5 mL portions were withdrawn at different reaction times and transferred into a Millipore ultrafiltration unit with a molar mass cut-off of 10000 D. After centrifugation with a Hermle Z 320K table centrifuge (5 min, 3000 rpm, 25 °C), the absorption spectrum of the filtrate containing aniline was measured with a NanoDrop ND1000 spectrometer. Analysis of the system before initiating the reaction was taken as reference.
2.10. ATR-FTIR measurements
Attenuated total reflectance-Fourier transformed infrared (ATR-FTIR) spectra of PANI were recorded with an Alpha instrument from Bruker. Around 6 mg sample was used. PANI was isolated by first adding acetone to the reaction system to precipitate the formed PANI. The precipitate was collected by centrifugation using a Hermle Z 320K table centrifuge, followed by washing of the precipitate first with acetone/water (1:1) and then with pure acetone. The sample was then dried in vacuum overnight.
3. Results and discussion
3.1. AOT vesicles
AOT is a widely used anionic amphiphile with two short branched hydrophobic tails (Scheme 2). The most popular application of AOT is as surfactant for the formation of reverse micelles in organic solvents like isooctane.29 AOT is actually thesurfactant for reverse micelle formation due to its molecular geometry which is in a first approximation like a truncated inverted cone, prone to form inverted structures in an apolar organic solvent. In dilute aqueous solution, AOT may aggregate into micelles or vesicles, depending on the experimental conditions. In the present work, we took advantage of this latter fact that AOT vesicle formation indeed can occur at low AOT concentration. For a better understanding of the AOT vesicle system which we later used as chemical structure-controlling template for the polymerization reaction, literature data and our own observations on the aggregation behavior of AOT in dilute aqueous solution are summarized in this section.In dilute aqueous solutions prepared with deionized water—without added salt—AOT forms micelles above the critical concentration for micelle formation (cmc). The recently determined cmc value for AOT in deionized water at 25 °C varies between 0.63 mM30 and 2.4–2.7 mM.31 In 1987, Ghosh and Miller reported that dispersing AOT in an aqueous solution at concentrations below 1 wt% (corresponding to 23 mM AOT) at 30 °C led to the formation of micelles (isotropic L1 phase), which were found to coexist with a lamellar liquid crystalline phase (dispersed Lα phase, i.e.vesicles) if salt in the form of NaCl was added.32 Regions of coexistence of L1 and Lα phases appeared if the AOT concentration was above about 0.25 wt% (= 5.6 mM AOT) and if the aqueous solution contained NaCl at a concentration between about 0.1 wt% (= 17 mM) and about 1.4 wt% (= 239 mM).30,32 The driving force for the formation of vesicles in the presence of salt is the screening of the electrostatic repulsion between adjacent sulfonate head groups leading to a transformation of AOT micelles into AOT vesicles.33 Originally, this transformation was found to occur at the reported L1 + Lα phase boundary, at about 5.6 mM for 17–239 mM NaCl.30,32 This seems to be incorrect since vesicle formation was also observed in the region of the phase diagram which was originally assigned to the pure L1 phase.30 This means that the originally reported cmc values in the presence of NaCl probably represent values for the critical concentration for the formation of vesicles (cvc).20 At 4.7 mM NaCl, the cvc for AOT was found to be 7.8 mM at 30 °C.34 At NaCl concentrations above 25 mM, the cvc values were reported to be below 1 mM AOT, with a tendency to decrease with increasing NaCl concentration (up to 170 mM).30 All these literature data clearly showed that AOT vesicle formation in dilute aqueous solution occurs if the aqueous solution contains added NaCl.
In the present work, we did not use NaCl but we dispersed AOT in 0.1 M phosphate solution (pH = 4.3), which was found to be optimal for the HRP-catalyzed polymerization of aniline,6–9 and we wondered whether AOT vesicle formation also occurs under these conditions. Since this was the case, the vesicles were used later on as templates for the polymerization reaction. The AOT vesicle formation was studied as follows. In a first set of experiments, the transmission (expressed as absorbance at 400 nm (arbitrarily chosen)) was measured as a function of AOT concentration (Fig. 1). A decrease in light transmission at 400 nm was taken as the indication of the presence of vesicles (large aggregates) which scatter visible light. In control measurements, solutions at different AOT concentrations in deionized water and in methanol were also analyzed in the same way. The results for 0.1 M phosphate solution (curve a), for deionized water (curve b) and for methanol (curve c) are shown in Fig. 1. Since the hydrophilic head group and the hydrophobic tails of AOT molecules have a good solubility in methanol, it is expected that no large aggregates form in methanol solution, which is consistent with the measurements (no turbidity up to at least 6 mM AOT (curve c in Fig. 1). In deionized water, the solutions also did not scatter visible light (curve b in Fig. 1), indicating again that no large aggregates formed up to 6 mM AOT. Therefore, there was no indication of vesicle formation in deionized water in the concentration range investigated, in agreement with literature.30–33 Whether micelles formed in deionized water below 6 mM AOT was not of interest and also could not be determined by these simple measurements since the micelles would be too small to scatter visible light. The determined aggregation number of AOT micelles is 29, see Grillo et al.33 In 0.1 M phosphate solution (pH = 4.3), the samples were turbid above about 0.4 mM AOT (curve a in Fig. 1), indicative of the formation of large aggregates (vesicles). The presence of vesicles was confirmed by cryo-TEM measurements (Fig. 2). For this electron microscopy analysis, the sample was homogenized with the freezing–thawing extrusion method,9,26 see Section 2.2. Even if the few small dark dots present in the images—most likely arising from a contamination by small ice crystallites—would be AOT micelles, they would amount to less than 5% of the AOT present as vesicles (as judged from samples containing 6 mM AOT). There was no indication of the presence of cylindrical micelles or flat bilayered sheets.
 | ||
| Fig. 1 Determination of the critical concentration for vesicle formation, cvc, of AOT by measuring the turbidity of AOT solutions and suspensions as a function of AOT concentration. The optical density was measured at λ = 400 nm (OD400), path length = 1 cm. (a, filled square) AOT in 0.1 M H2PO4− (pH = 4.3); (b, open triangle) AOT in water; and (c, open circle) AOT in methanol. | ||
![Cryo-TEM images of AOT vesicles, prepared by the freezing–thawing extrusion method (see Materials and methods) in 0.1 M H2PO4−, pH = 4.3. [AOT] = 20 mM (a) and 6.0 mM (b). Length of the bar: 100 nm.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f2.gif) | ||
| Fig. 2 Cryo-TEM images of AOT vesicles, prepared by the freezing–thawing extrusion method (see Materials and methods) in 0.1 M H2PO4−, pH = 4.3. [AOT] = 20 mM (a) and 6.0 mM (b). Length of the bar: 100 nm. | ||
The AOT vesicles obtained by extrusion were rather homogeneous with respect to size and most of the vesicles were unilamellar (Fig. 2). The average size of the vesicles determined by dynamic light scattering (DLS) measurements was 72 nm immediately after the preparation with an increase in size upon storage at room temperature up to 108 nm after 7 days (Fig. 3). Such vesicle size increase upon storage was also observed for SDBS/decanoic acid (1:1) vesicles,9 indicating (i) that the average size to which the vesicles were forced by extrusion was not the thermodynamically most stable size and (ii) that the activation energy for vesicle fusion was low enough so that significant vesicle fusion occurred at room temperature within a few days of storage.
![Dynamic light scattering measurements of AOT vesicles, prepared by the freezing–thawing extrusion method (see Materials and methods) and stored at room temperature. [AOT] = 1 mM, 0.1 M H2PO4−, pH = 4.3.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f3.gif) | ||
| Fig. 3 Dynamic light scattering measurements of AOT vesicles, prepared by the freezing–thawing extrusion method (see Materials and methods) and stored at room temperature. [AOT] = 1 mM, 0.1 M H2PO4−, pH = 4.3. | ||
3.2. Optimization of the reaction system
Like in the HRP/H2O2 systems studied previously with SDBS/decanoic acid (1:1) vesicles as templates,9 the absorption maximum of the emeraldine salt form of PANI under optimal reaction conditions in the presence of AOT vesicles was found to be at about 1000 nm. This agrees with the observations made with sulfonated polystyrene as template7 or with micellar templates formed from surfactants which have a sulfonate head group (SDBS or sodium dodecyldiphenyloxidedisulfonate).8
The AOT vesicle reaction system was optimized by varying the AOT concentration, the sodium dihydrogen phosphate concentration and the temperature.
![HRP-catalyzed polymerization of aniline in the presence of AOT vesicle templates. Variation of the ratio A1000 nm/A550 nm as a function of AOT concentration at pH = 4.3 (0.1 M H2PO4−), [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, t = 1 h, T ≈ 25 °C.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f4.gif) | ||
| Fig. 4 HRP-catalyzed polymerization of aniline in the presence of AOT vesicle templates. Variation of the ratio A1000 nm/A550 nm as a function of AOT concentration at pH = 4.3 (0.1 M H2PO4−), [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, t = 1 h, T ≈ 25 °C. | ||
As the AOT concentration increased from 0.3 mM to 15 mM, A1000 nm/A550 nm first increased and reached a maximum value at 1 mM AOT, then decreased. This observation can be explained as follows: at pH = 4.3, about 67% of the aniline molecules are protonated and have a positive charge which may efficiently complex with the negatively charged AOT vesicles prior to the reaction (pKa (anilinium) = 4.6). If the AOT concentration is low (as compared to the aniline concentration), most of the aniline molecules are expected to freely move in the solution before and during the polymerization, leading to the formation of more branched structures since there is no direct control of the polymer chain elongation by the vesicle template. At too high AOT concentration, the local concentration of aniline on the vesicle surface decreases which may result in the formation of increased amounts of short polymer chains. This would explain the decrease in A1000 nm/A550 nm at high AOT concentration. In any case, the optimum AOT concentration at [aniline] = 1.33 mM was found to be 1 mM (1 mM H2O2, 0.1 M H2PO4−, pH = 4.3), see Fig. 4.
![Effect of added NaH2PO4 concentration on the HRP-catalyzed polymerization of aniline in the presence of AOT vesicles. The absorbance at 1000 nm of the reaction solution was measured 24 h after the start of the reaction. [AOT] = 1 mM; [aniline] = 1.33 mM; [HRP] = 25 µg mL−1; [H2O2] = 1 mM; path length = 0.1 cm; T ≈ 25 °C.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f5.gif) | ||
| Fig. 5 Effect of added NaH2PO4 concentration on the HRP-catalyzed polymerization of aniline in the presence of AOT vesicles. The absorbance at 1000 nm of the reaction solution was measured 24 h after the start of the reaction. [AOT] = 1 mM; [aniline] = 1.33 mM; [HRP] = 25 µg mL−1; [H2O2] = 1 mM; path length = 0.1 cm; T ≈ 25 °C. | ||
These observations can be understood on the basis of the following consideration. Sodium ions compete with anilinium ions and H3O+ and with (protonated) oligoaniline chains for binding to the vesicle surface. It is therefore likely that the amount of bound anilinium ions at constant pH is decreased at high NaH2PO4 concentration. As a consequence, the AOT vesicles will lose their template effect. Furthermore, since protons are released from the amino groups during polymerization,1d,35 there may be a significant decrease of the pH value if the buffering capacity of the phosphate solution at low phosphate concentration is insufficient. If the pH value drops to a too low value, the HRP may become less active, and the relative amount of unprotonated aniline molecules which can be oxidized by HRP decreases at low pH. Although the observed effect of NaH2POP4 concentration requires further investigations, it seems that a concentration of about 0.1 M NaH2PO4 is optimal.
![HRP-catalyzed synthesis of PANI in the presence of AOT vesicles. VIS/NIR spectra of PANI obtained after 24 h at two different temperatures: (a, solid line) at T = 7 °C and (b, dashed line) at T = 25 °C. [AOT] = 1 mM, [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, t = 24 h pH = 4.3 (0.1 M H2PO4−), and path length = 0.1 cm.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f6.gif) | ||
| Fig. 6 HRP-catalyzed synthesis of PANI in the presence of AOT vesicles. VIS/NIR spectra of PANI obtained after 24 h at two different temperatures: (a, solid line) at T = 7 °C and (b, dashed line) at T = 25 °C. [AOT] = 1 mM, [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, t = 24 h pH = 4.3 (0.1 M H2PO4−), and path length = 0.1 cm. | ||
The absorption bands at about 1000 nm and about 420 nm were more intense when the reaction was carried out at 7 °C, as compared to 25 °C. Absorption in both regions of the spectrum can be attributed to delocalized electrons of the radical cations (separated and delocalized polarons) that are characteristic for the conductive emeraldine salt form of PANI.1f,19–21,36 The higher band intensities around 1000 nm and 420 nm of PANI obtained at 7 °C as compared to room temperature may indicate that the PANI obtained at the lower reaction temperature contained more structural units with extended conjugation, possibly longer chains, if compared to the PANI obtained at room temperature. This latter interpretation would be consistent with reports on the temperature dependency of the formation of PANI by purely chemical, non-enzymatic methods.37 Although in that case, the experimental conditions were very different from the conditions used in our system, a lower reaction temperature led to the formation of longer polymer chains.37 The possibility that the chains obtained at 7 °C had a more extended coil-like conformation19 as compared to the chains obtained at 25 °C would be consistent with the observed decreased absorbance at ∼300 nm (π–π* transition).19 A decrease in band intensity at ∼300 nm—with an increase in the NIR region of the spectrum (above ∼800 nm)—was observed if compact coils of the emeraldine salt form of PANI were converted into extended coils by changing the solvent.19
The higher absorbance at ∼420 nm and ∼1000 nm observed at 7 °C was not related to a higher reaction yield at 7 °C, as compared to 25 °C, see Fig. 7. Independent of whether the polymerization reaction was carried out at 7 °C or 25 °C, the remaining aniline in the reaction system quickly dropped to about 40% within the first hour and to 30% after 24 h, and then stayed constant for at least another day. This reveals that the HRP-catalyzed polymerization of aniline under the experimental conditions used is a fast process and the consumption of aniline in the presence of AOT vesicles mainly occurred during the first hour.
![Change in the percentage of remaining aniline in the reaction systems during the reaction carried out at two different reaction temperatures, at T = 7 °C (filled circle) and at 25 °C (open square). The reaction conditions were: [AOT] = 1 mM, [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, in pH = 4.3 (0.1 M H2PO4−).](/image/article/2011/SM/c0sm00599a/c0sm00599a-f7.gif) | ||
| Fig. 7 Change in the percentage of remaining aniline in the reaction systems during the reaction carried out at two different reaction temperatures, at T = 7 °C (filled circle) and at 25 °C (open square). The reaction conditions were: [AOT] = 1 mM, [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, in pH = 4.3 (0.1 M H2PO4−). | ||
3.3. Interaction of HRP with AOT vesicles
![Effect of HRP on the fluorescence of TNS in the presence of AOT vesicles (1 mM AOT) at pH = 4.3 (0.1 M H2PO4−). [TNS] = 3.2 µM, λex = 320 nm, λem = 422 nm, T = 25 °C. The relative fluorescence intensity of TNS is plotted as a function of HRP concentration.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f8.gif) | ||
| Fig. 8 Effect of HRP on the fluorescence of TNS in the presence of AOT vesicles (1 mM AOT) at pH = 4.3 (0.1 M H2PO4−). [TNS] = 3.2 µM, λex = 320 nm, λem = 422 nm, T = 25 °C. The relative fluorescence intensity of TNS is plotted as a function of HRP concentration. | ||
:1) vesicles,9 where the presence of vesicles led to a measurable decrease of the ellipticity of HRP in the Soret band region.9 Based on the CD analysis it can be concluded that the AOT vesicles (1 mM) did not affect the structure of HRP at pH = 4.3 significantly, despite there being evidence that the enzyme interacts with the vesicles (Fig. 8). Enzyme binding to the vesicles appears to occur away from the active site, without altering the heme group environment.
![Effect of AOT vesicles on the CD spectrum of horseradish peroxidase C (12 µM = 480 µg mL−1) in (a) the far UV region and (b) the Soret band region of the spectrum. [AOT] = 1 mM, 0.1 M H2PO4−, pH = 4.3, T = 25 °C. (a) Path length = 0.1 cm and (b) path length = 0.5 cm. Solid line: in the presence of AOT vesicles and dashed line: in phosphate solution (no vesicles).](/image/article/2011/SM/c0sm00599a/c0sm00599a-f9.gif) | ||
| Fig. 9 Effect of AOT vesicles on the CD spectrum of horseradish peroxidase C (12 µM = 480 µg mL−1) in (a) the far UV region and (b) the Soret band region of the spectrum. [AOT] = 1 mM, 0.1 M H2PO4−, pH = 4.3, T = 25 °C. (a) Path length = 0.1 cm and (b) path length = 0.5 cm. Solid line: in the presence of AOT vesicles and dashed line: in phosphate solution (no vesicles). | ||
:1) vesicles ([SDBS] = [decanoic acid] = 0.665 mM) as templates are also shown in Fig. 10.
![Stability of HRP in different reaction systems prepared at pH = 4.3 (0.1 M H2PO4−) in the absence of vesicles, in the presence of AOT vesicles and in the presence of SDBS/decanoic acid (1 : 1) vesicles. HRP (25 µg mL−1), aniline (1.33 mM) and the vesicles were incubated for a certain period of time, followed by the addition of H2O2 (1.0 mM in the reaction mixture) to start the reaction. After 1 h, the VIS/NIR spectrum was recorded and the value of absorbance at λ = 1000 nm was taken as a measure for the activity of the enzyme, plotted as a function of time (path length = 0.1 cm). The values given are relative values, normalized by the absorbance obtained without incubation. Filled bar: in the presence of AOT vesicles ([AOT] = 1.0 mM); dashed bar: in the absence of vesicles; and empty bar: in the presence of SDBS/decanoic acid (1 : 1) vesicles ([SDBS] = [decanoic acid] = 0.665 mM).](/image/article/2011/SM/c0sm00599a/c0sm00599a-f10.gif) | ||
| Fig. 10 Stability of HRP in different reaction systems prepared at pH = 4.3 (0.1 M H2PO4−) in the absence of vesicles, in the presence of AOT vesicles and in the presence of SDBS/decanoic acid (1:1) vesicles. HRP (25 µg mL−1), aniline (1.33 mM) and the vesicles were incubated for a certain period of time, followed by the addition of H2O2 (1.0 mM in the reaction mixture) to start the reaction. After 1 h, the VIS/NIR spectrum was recorded and the value of absorbance at λ = 1000 nm was taken as a measure for the activity of the enzyme, plotted as a function of time (path length = 0.1 cm). The values given are relative values, normalized by the absorbance obtained without incubation. Filled bar: in the presence of AOT vesicles ([AOT] = 1.0 mM); dashed bar: in the absence of vesicles; and empty bar: in the presence of SDBS/decanoic acid (1:1) vesicles ([SDBS] = [decanoic acid] = 0.665 mM). | ||
After 3 h pre-incubation, HRP showed similar activity in phosphate solution and in the presence of AOT vesicles or in the presence of SDBS/decanoic acid (1:1) vesicles. In all three cases, the activity was almost the same as without pre-incubation (set to 100% in Fig. 10). If the HRP was pre-incubated for 20 h at 25 °C, the enzyme in the presence of AOT vesicles was still fully active like without incubation or after incubation in phosphate solution without vesicles. In contrast, the activity of HRP in the presence of SDBS/decanoic acid (1:1) vesicles decreased to about 70%. Based on these results it is evident that HRP remains more stable in the presence of AOT vesicles as compared to the SDBS/decanoic acid (1:1) vesicles used previously.9 In this respect, the AOT vesicle system is a better template system than the SDBS/decanoic acid (1:1) system, which in turn was already shown to be better than SDBS micelles.9
3.4. Interaction of aniline with AOT vesicles
:1) vesicles.9 This fully supports the view that aniline molecules do not have strong interactions with AOT vesicles under the conditions used, which is in clear contrast to the behavior of HRP (Fig. 8). Aniline probably just loosely associates on the vesicle surface. It may, however, also be that possible aniline–AOT interactions are not sensed by the vesicle embedded membrane probe TNS. Therefore, NMR measurements were also carried out, see below.
![Effect of aniline on the fluorescence of TNS in the presence of AOT vesicles (1 mM AOT) at pH = 4.3 (0.1 M H2PO4−). [TNS] = 1 µM, λex = 320 nm, λem = 422 nm, T = 25 °C. The relative fluorescence intensity of TNS is plotted as a function of aniline concentration.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f11.gif)
![700.13 MHz 1H–1H NOESY spectrum of a mixture of aniline and AOT vesicles in deuterated phosphate solution. [AOT] = [aniline] = 8 mM, pD = 3.9, T = 25 °C. Strong intramolecular NOE cross-peaks are indicated with the solid squares. These NOEs originate from proton interactions in aniline and from proton interactions in AOT, respectively. Weak intermolecular NOE crosspeaks are found between resonances of aniline and the aliphatic chains of AOT, as indicated with the dashed square.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f12.gif) | ||
| Fig. 12 700.13 MHz 1H–1H NOESY spectrum of a mixture of aniline and AOT vesicles in deuterated phosphate solution. [AOT] = [aniline] = 8 mM, pD = 3.9, T = 25 °C. Strong intramolecular NOE cross-peaks are indicated with the solid squares. These NOEs originate from proton interactions in aniline and from proton interactions in AOT, respectively. Weak intermolecular NOE crosspeaks are found between resonances of aniline and the aliphatic chains of AOT, as indicated with the dashed square. | ||
3.5. Analysis of the reaction system during polymerization
:1) vesicle system, the VIS/NIR data may indicate that during the early phase of the reaction the reaction products obtained were overoxidized (peak at 750 nm). The typical spectrum of the emeraldine salt form with its characteristic absorption in the NIR region of the spectrum and at 400 nm only developed in the second phase of the reaction, when the intensity of the peak at 750 nm continuously decreased. Our interpretation about the formation of overoxidized PANI at the early stage of the enzymatic polymerization reaction would be in agreement with reports on the purely chemical, non-enzymatic polymerization of aniline with (NH4)2S2O8 in 1.0 M HCl.39 In that case, the fully oxidized pernigraniline form of PANI was found to form first before further reaction leads to the formation of the half oxidized emeraldine form.39 There are, however, two other points to consider. First, a similar transition with a decrease of the band intensity at 750 nm and an increase in the band intensity in the NIR region of the spectrum (at λ > 800 nm) was observed when coiled emeraldine salt conformations transformed into expanded coil-like conformations,19 apparently without any externally triggered chemical reactions occurring. Second, Fig. 7 shows that after 1 h of reaction the concentration of non-reacted aniline remained almost constant; the monomer integration into the polymer chains apparently was already almost completed. Experiments are now in progress with the aim of hopefully clarifying the situation at the early stage of the reaction.
![Time dependent changes of the VIS/NIR absorption spectrum during the HRP-catalyzed polymerization of aniline in the presence of AOT vesicles carried out at T = 7 °C. [AOT] = 1 mM; [aniline] = 1.33 mM; [HRP] = 25 µg mL−1; [H2O2] = 1 mM; pH = 4.3 (0.1 M H2PO4−), path length: 0.2 cm. (a) Absorption spectrum of the reaction system as a function of reaction time; the first spectrum was recorded 10 min after the start of the reaction (after the addition of H2O2), the following spectra were recorded in intervals of 20 min; the arrows indicate direction of the changes of the intensities with time. (b) Changes of the absorbance at λ = 1000 nm, 750 nm and 400 nm with reaction time.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f13.gif) | ||
| Fig. 13 Time dependent changes of the VIS/NIR absorption spectrum during the HRP-catalyzed polymerization of aniline in the presence of AOT vesicles carried out at T = 7 °C. [AOT] = 1 mM; [aniline] = 1.33 mM; [HRP] = 25 µg mL−1; [H2O2] = 1 mM; pH = 4.3 (0.1 M H2PO4−), path length: 0.2 cm. (a) Absorption spectrum of the reaction system as a function of reaction time; the first spectrum was recorded 10 min after the start of the reaction (after the addition of H2O2), the following spectra were recorded in intervals of 20 min; the arrows indicate direction of the changes of the intensities with time. (b) Changes of the absorbance at λ = 1000 nm, 750 nm and 400 nm with reaction time. | ||
Sections of the 1H NMR spectrum of the reaction system recorded at different times after the start of the reaction are presented in Fig. 14, together with a reference spectrum of the reaction solution before the start of the reaction, i.e. before the addition of H2O2 (Fig. 14). Three obvious time-dependent changes could be identified in comparison with the reference spectrum: (i) the signal intensity of the aromatic protons at 7.0–7.3 ppm showed a significant decrease one hour after the start of the reaction; (ii) a clear peak shift to higher frequency of the meta-, para- and ortho-protons of aniline could be observed. After one hour, the signal intensity and the position of the peaks corresponding to the meta-, para- and ortho-protons remained constant; and (iii) the NMR signals attributed to the AOT molecules (at δ ≈ 1 ppm) and the aniline resonances at 7.0–7.3 ppm became broader. No signals originating from reaction products could be detected, despite the fact that the integral of the aniline protons between 7.0 and 7.3 ppm after 4.5 h of reaction dropped to ∼45% of the integral measured at the beginning of the reaction. This means that ∼55% of the initial aniline reacted. This is comparable with the independently determined amount of unreacted aniline left at reaction equilibrium, see Fig. 7.
![HRP-catalyzed polymerization of aniline in the presence of AOT vesicles. 1H NMR spectrum of the reaction solution recorded at different time. (a) Between 7.00 and 7.35 ppm and (b) between 0.00 and 2.00 ppm. From bottom to top: in the absence of H2O2 (no reaction); 1 h after the addition of H2O2 to start the reaction; 2 h after the start of the reaction; 4.5 h after the start of the reaction. [AOT] = 1 mM; [aniline] = 1.33 mM; [H2O2] = 1 mM, pD = 3.9 (0.1 M D2PO4−).](/image/article/2011/SM/c0sm00599a/c0sm00599a-f14.gif) | ||
| Fig. 14 HRP-catalyzed polymerization of aniline in the presence of AOT vesicles. 1H NMR spectrum of the reaction solution recorded at different time. (a) Between 7.00 and 7.35 ppm and (b) between 0.00 and 2.00 ppm. From bottom to top: in the absence of H2O2 (no reaction); 1 h after the addition of H2O2 to start the reaction; 2 h after the start of the reaction; 4.5 h after the start of the reaction. [AOT] = 1 mM; [aniline] = 1.33 mM; [H2O2] = 1 mM, pD = 3.9 (0.1 M D2PO4−). | ||
The absence of NMR signals arising from the reaction product(s) is in agreement with the formation of delocalized unpaired (paramagnetic) electrons, which lead to broad signals that are not measurable. Such delocalized unpaired electrons are expected to be present in the emeraldine salt form of PANI. Therefore, this interpretation of the NMR measurements would be in agreement with the interpretation of the VIS/NIR spectrum in the sense that most of the reaction products obtained had the expected chemical structure of the emeraldine salt form of PANI (Scheme 1). An alternative explanation for the absence of NMR signals from the polymeric product(s) is the polymer binding to the vesicles which leads to a slowing down of the molecular motions and consequently to a broadening of the signals. If the signals become too broad they are not detectable.
The absence of 1H NMR signals for the reaction products is in agreement with literature data from a 1H NMR study of the chemical synthesis of PANI with Na2S2O8.40 In that case, the reaction products were also not visible in a 1H NMR spectrum,40 independent of whether the products precipitated—absence of signals from insoluble polymers—or whether they were kept as stable dispersion with the help of SDBS.40
The broadening of the AOT 1H NMR signals at ∼1 ppm and of the aniline resonances at 7.0–7.3 ppm is probably due to intermolecular paramagnetic dipolar relaxation caused by the presence of unpaired electrons. The peak shift of the aromatic protons of aniline in the spectrum recorded 1 h, 2 h and 4.5 h after H2O2 addition (Fig. 14) may indicate that the remaining, unreacted, aniline molecules are no more so close to the vesicle surface. This possibility is based on the fact that the chemical shifts of the aniline protons in aqueous solution at pH = 4.3 (in the absence of vesicles) are similar to the chemical shifts of aniline measured after 1 h, 2 h, or 4.5 h of reaction (Fig. 14). This would mean that the unreacted aniline monomers are displaced from the vesicle surface by the PANI formed which itself binds to the vesicles. Further experiments are certainly needed to hopefully clarify this point.
![Cryo-TEM images of the AOT vesicular reaction system after reaching reaction equilibrium. The polymerization reaction was carried out at T = 7 °C under the following conditions: [AOT] = 3 mM, [aniline] = 4 mM, [HRP] = 75 µg mL−1, [H2O2] = 3 mM, pH = 4.3 (0.1 M H2PO4−). The reaction time was 15 h. The vesicles were prepared by the freezing–thawing extrusion method (see Materials and methods). Length of the bar in both images, a and b, is 100 nm.](/image/article/2011/SM/c0sm00599a/c0sm00599a-f15.gif) | ||
| Fig. 15 Cryo-TEM images of the AOT vesicular reaction system after reaching reaction equilibrium. The polymerization reaction was carried out at T = 7 °C under the following conditions: [AOT] = 3 mM, [aniline] = 4 mM, [HRP] = 75 µg mL−1, [H2O2] = 3 mM, pH = 4.3 (0.1 M H2PO4−). The reaction time was 15 h. The vesicles were prepared by the freezing–thawing extrusion method (see Materials and methods). Length of the bar in both images, a and b, is 100 nm. | ||
3.6. Characterization of the PANI product obtained by FTIR spectroscopy
The reaction product obtained from aniline with HRP/H2O2 in the presence of AOT vesicles at 7 °C was isolated from the reaction mixture (see Section 2.10) and then analyzed by ATR-FTIR spectroscopy (Fig. 16). The individual bands in the FTIR spectrum can be assigned as follows. The two distinct peaks at 1595 cm−1 and 1501 cm−1 are characteristic bands of the vibrational modes of the quinoid units (C–C stretching at 1595 cm−1) and of the benzenoid units (C–C stretching at 1501 cm−1) of the emeraldine form of PANI1d,f,9,23a,41 The absorption band at 1595 cm−1 supports the presence of aniline units in the PANI obtained that are connected via a head-to-tail monomer coupling during polymerization.1d,42 The strong absorptions at 1306 cm−1 and 1160 cm−1 are caused by the C–N stretching of aromatic amines and by the C–H in plane deformation vibration, respectively.1d,f,9,41,42 The C–H out-of-plane bending mode located at 819 cm−1, which is characteristic for para-disubstituted aromatic rings,1d,f,9 is clearly observed. The band at 1248 cm−1 can be assigned to the C–N+˙ stretching in the polaron form of the emeraldine salt of PANI.41 Based on the literature,1d,41 the stretching vibration of –NH+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) in the bipolaron structure of the emeraldine salt form of PANI should be at 1146 cm−1 which is not clearly evident in the spectrum shown in Fig. 16. The band—if present—may be localized below the peak with maximum absorbance at 1160 cm−1. The weak bands located at 695 cm−1 and 749 cm−1 can be assigned to C–H out-of-plane deformation vibrations of monosubstituted aromatic rings.1f The presence of a band at ∼690 cm−1 has also been taken as an indication of ortho-disubstituted aromatic rings within the PANI chains.1d,9,43 Furthermore, the band at 749 cm−1 may also indicate the presence of ortho-coupled units.44 The small peak at ∼860 cm−1 may indicate the presence of some branched units,1d,9 occurring through an ortho-coupling of the aniline molecules during polymerization. If the intensities of the peaks which may arise from ortho-coupled aniline units (at 875 cm−1, 795 cm−1 and 695 cm−1) are compared with the intensities of the band characteristic for para-coupled aniline units (819 cm−1), it seems that most of the PANI obtained was linear with para-coupled aniline chains, as expected for the emeraldine form of PANI.
in the bipolaron structure of the emeraldine salt form of PANI should be at 1146 cm−1 which is not clearly evident in the spectrum shown in Fig. 16. The band—if present—may be localized below the peak with maximum absorbance at 1160 cm−1. The weak bands located at 695 cm−1 and 749 cm−1 can be assigned to C–H out-of-plane deformation vibrations of monosubstituted aromatic rings.1f The presence of a band at ∼690 cm−1 has also been taken as an indication of ortho-disubstituted aromatic rings within the PANI chains.1d,9,43 Furthermore, the band at 749 cm−1 may also indicate the presence of ortho-coupled units.44 The small peak at ∼860 cm−1 may indicate the presence of some branched units,1d,9 occurring through an ortho-coupling of the aniline molecules during polymerization. If the intensities of the peaks which may arise from ortho-coupled aniline units (at 875 cm−1, 795 cm−1 and 695 cm−1) are compared with the intensities of the band characteristic for para-coupled aniline units (819 cm−1), it seems that most of the PANI obtained was linear with para-coupled aniline chains, as expected for the emeraldine form of PANI.
![ATR-FTIR spectrum of PANI synthesized at 7 °C in the presence of AOT vesicles and isolated by using the acetone precipitation method (see Materials and methods). The reaction conditions were: [AOT] = 1 mM, [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, t = 24 h and pH = 4.3 (0.1 M H2PO4−).](/image/article/2011/SM/c0sm00599a/c0sm00599a-f16.gif) | ||
| Fig. 16 ATR-FTIR spectrum of PANI synthesized at 7 °C in the presence of AOT vesicles and isolated by using the acetone precipitation method (see Materials and methods). The reaction conditions were: [AOT] = 1 mM, [aniline] = 1.33 mM, [HRP] = 25 µg mL−1, [H2O2] = 1 mM, t = 24 h and pH = 4.3 (0.1 M H2PO4−). | ||
Ortho-coupling of aniline units, particularly occurring in chemical polymerizations at the early stage of the reaction,2b may lead to the formation of phenazine units. Typical band positions for vibrations of substituted phenazines are expected at 1623 cm−1, 1414 cm−1, 1208 cm−1, 1144 cm−1, 1136 cm−1, and 1108 cm−1.1f Since there was no clear absorption at these frequencies (see Fig. 16), the presence of large amounts of phenazine units in the PANI obtained can be excluded.
The assignment of the bands observed at 1072 cm−1 and 933 cm−1 is not clear at the moment. Although AOT has a strong band at ∼1050 cm−1 (SO stretching), the other characteristic band of AOT at ∼1730 cm−1 (CO stretching) is missing. The FTIR spectrum of the PANI obtained indicates that AOT could be removed from the reaction product quite efficiently, in contrast to the case of the SDBS/decanoic acid (1:1) vesicles used previously,9 where complete SDBS removal was not possible.9 In this respect, there is another advantage of the AOT vesicles over the previously used system.
4. Conclusions
This is the second time that experimental data on the use of vesicles as chemical structure-controlling templates for enzymatic polymerization reactions are presented. The reaction investigated was the HRP/H2O2-triggered polymerization of aniline at pH = 4.3 in 0.1 M H2PO4− solution and the vesicles used were formed from AOT. It was shown that AOT vesicles have clear advantages if compared with the SDBS/decanoic acid (1:1) vesicle system used previously.9 First, the enzyme, HRP, is more stable if incubated together with the AOT vesicles as compared to the SDBS/decanoic acid (1:1) vesicles. Second, the AOT vesicles are also stable at a temperature below 10 °C which is not the case for the SDBS/decanoic acid (1:1) vesicles.9 Since the reaction temperature has an influence on the conjugation length and/or the conformation of the PANI obtained (Fig. 6), the AOT system allows further studies of this point. Although several aspects of the entire reaction need to be elaborated further, the first results obtained indicate that the polymerization of aniline leads to a stable aqueous suspension which is composed of vesicles which are coated with the conductive emeraldine salt form of PANI. Compared to polymers and micelles as templates for the HRP/H2O2-catalyzed polymerization of aniline,5–8 the vesicle system is different since it allows the preparation of hollow polymeric capsules, which is not possible with polymers or micelles as templates. Hollow capsules of conductive polymers have recently attracted the interest of several groups for various potential applications, i.e. for the development of sensor or delivery systems.45
Whether these vesicle polymer capsules can be processed, for example to form thin films, remains to be seen. Furthermore, the applicability of vesicles as chemical structure-controlling templates for other enzymatic polymerization reactions needs to be clarified. Based on a more general consideration,4 there seems to be a restriction for using chemical structure-controlling templates (polymers, micelles, vesicles, etc.) for those enzymatic reactions in which the growth of the polymer chain does not occur at the active site of the enzyme, similarly to the final steps in the biosynthesis of lignin.4 Further investigations toward a better understanding of the reaction mechanism in the vesicle system are currently underway.
Acknowledgements
The authors thank Dr Martin Willeke, Department of Materials, ETH Zürich, for many discussions and for the critical and careful reading of the manuscript. We acknowledge EMEZ (Electron Microscopy Center, ETH Zürich), especially Peter Tittmann and Dr Roger Wepf, for technical support. The financial support by the Swiss National Science Foundation (200021-111696 and 200020-124690) is highly appreciated.Notes and references
- (a) A. G. MacDiarmid and A. J. Epstein, Faraday Discuss. Chem. Soc., 1989, 88, 317–332 RSC; (b) E. M. Geniès, A. Boyle, M. Lapkowski and C. Tsintavis, Synth. Met., 1990, 36, 139–182 CrossRef CAS; (c) J. Stejskal, P. Kratochvil and A. D. Jenkins, Polymer, 1996, 37, 367–369 CrossRef CAS; (d) M. Trchová, I. Šeděnková, E. N. Konyushenko, J. Stejskal, P. Holler and G. Cirić-Marjanović, J. Phys. Chem. B, 2006, 110, 9461–9468 CrossRef CAS; (e) J. Křiž, L. Starovoytova, M. Trchová, E. N. Konyushenko and J. Stejskal, J. Phys. Chem. B, 2009, 113, 6666–6673 CrossRef CAS; (f) E. Dimitrieva, Y. Harima and L. Dunsch, J. Phys. Chem. B, 2009, 113, 16131–16141 CrossRef; (g) S. Bhadra, D. Khastgir, N. K. Singh and J. H. Lee, Prog. Polym. Sci., 2009, 34, 783–810 CrossRef CAS.
- (a) Y. Cao, A. Andreatta, A. J. Heeger and P. Smith, Polymer, 1989, 30, 2305–2311 CrossRef CAS; (b) I. Sapurina and J. Stejskal, Polym. Int., 2008, 57, 1295–1325 CrossRef CAS.
- P. Xu, A. Singh and D. L. Kaplan, Adv. Polym. Sci., 2006, 194, 69–94 CAS.
- P. Walde and Z. Guo, Soft Matter, 2010 10.1039/c0sm00259c.
- L. A. Samuelson, A. Anagnostopoulos, K. S. Alva, J. Kumar and S. K. Tripathy, Macromolecules, 1998, 31, 4376–4378 CrossRef CAS.
- (a) W. Liu, A. L. Cholli, R. Nagarajan, J. Kumar, S. Tripathy, F. F. Bruno and L. A. Samuelson, J. Am. Chem. Soc., 1999, 121, 11345–11355 CrossRef CAS; (b) S. K. Sahoo, R. Nagarajan, S. Roy, L. A. Samuelson, J. Kumar and A. L. Cholli, Macromolecules, 2004, 37, 4130–4138 CrossRef CAS.
- W. Liu, J. Kumar, S. Tripathy, K. J. Senecal and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 71–78 CrossRef CAS.
- (a) W. Liu, J. Kumar, S. Tripathy and L. A. Samuelson, Langmuir, 2002, 18, 9696–9704 CrossRef CAS; (b) V. Rumbau, J. A. Pomposo, J. A. Alduncin, H. Grande, D. Mecerreyes and E. Ochoteco, Enzyme Microb. Technol., 2007, 40, 1412–1421 CrossRef CAS.
- Z. Guo, H. Rüegger, R. Kissner, T. Ishikawa, M. Willeke and P. Walde, Langmuir, 2009, 25, 11390–11405 CrossRef CAS.
- M. R. Nabid, M. Golbabaee, A. B. Moghaddam, A. R. Mahdavian and M. M. Amini, Polym. Compos., 2009, 30, 841–846 Search PubMed.
- J. P. Foreman and A. P. Monkman, J. Phys. Chem. A, 2003, 107, 7604–7610 CrossRef CAS.
- (a) Y. Cao, P. Smith and A. J. Heeger, Synth. Met., 1989, 32, 263–281 CrossRef CAS; (b) J. Stejskal, P. Kratochvil and N. Radhakrishnan, Synth. Met., 1993, 61, 225–231 CrossRef CAS; (c) W. S. Huang and A. G. MacDiarmid, Polymer, 1993, 34, 1833–1845 CrossRef CAS.
- A. J. Epstein, R. P. McCall, J. M. Grinder and A. G. MacDiarmid, in Spectroscopy of Advanced Materials, ed. R. J. H. Clark and R. E. Hester, John Wiley & Sons, Chichester, 1991, pp. 355–396 Search PubMed.
- M. Cochet, B. Corraze, S. Quillard, J. P. Buisson, S. Lefrant and G. Louarn, Synth. Met., 1997, 84, 757–758 CrossRef CAS.
- (a) R. Colle, P. Parruccini, A. Benassi and C. Cavazzoni, J. Phys. Chem. B, 2007, 111, 2800–2805 CrossRef CAS; (b) A. Varela-Álvarez and J. A. Sordo, J. Chem. Phys., 2008, 128, 174706 CrossRef.
- H. Zhekova, A. Tadjer, A. Ivanova, J. Petrova and N. Gospodinova, Int. J. Quantum Chem., 2007, 107, 1688–1706 CrossRef CAS.
- L. Cristofolini, M. P. Fontana, P. Camorani, T. Berzina and A. Nabok, Langmuir, 2010, 26, 5829–5835 CrossRef CAS.
- T. Lindfors and L. Harju, Synth. Met., 2008, 158, 233–241 CrossRef CAS.
- (a) A. G. MacDiarmid and A. J. Epstein, Synth. Met., 1994, 65, 103–116 CrossRef; (b) Y. Xia, J. M. Wiesinger, A. G. MacDiarmid and A. J. Epstein, Chem. Mater., 1995, 7, 443–445 CrossRef CAS.
- S. E. Moulton, P. C. Innis, L. A. P. Kane-Maguire, O. Ngamna and G. G. Wallace, Curr. Appl. Phys., 2004, 4, 402–406 CrossRef.
- (a) A. A. Nekrasov, V. F. Ivanov and A. V. Vannikov, J. Electroanal. Chem., 2000, 482, 11–17 CrossRef CAS; (b) A. A. Nekrasov, V. F. Ivanov and A. V. Vannikov, Electrochim. Acta, 2001, 46, 3301–3307 CrossRef CAS.
- C. H. Lim and Y. J. Yoo, Process Biochem. (Amsterdam, Neth.), 2000, 36, 233–241 Search PubMed.
- (a) G. Louarn, M. Lapkowski, S. Quillard, A. Pron, J. P. Buisson and S. Lefrant, J. Phys. Chem., 1996, 100, 6998–7006 CrossRef CAS; (b) I. Šeděnkova, M. Trchová, J. Stejskal and J. Box, Appl. Spectrosc., 2007, 61, 1153–1162 CrossRef CAS.
- Z. Ping, J. Chem. Soc., Faraday Trans., 1996, 92, 3063–3067 RSC.
- H. B. Dunford and J. S. Stillman, Coord. Chem. Rev., 1976, 19, 187–251 CrossRef.
- L. D. Mayer, M. J. Hope and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1986, 858, 161–168 CrossRef CAS.
- K. Chattopadhyay and S. Mazumdar, Biochemistry, 2000, 39, 263–270 CrossRef CAS.
- T. Namani, T. Ishikawa, K. Morigaki and P. Walde, Colloids Surf., B, 2007, 54, 118–123 CrossRef CAS.
- T. K. De and A. Maitra, Adv. Colloid Interface Sci., 1995, 59, 95–193 CrossRef CAS.
- N. Shahidzadeh, D. Bonn and J. Meunier, Europhys. Lett., 1997, 40, 459–464 CrossRef CAS.
- (a) A. Chatterjee, S. P. Moulink, S. K. Sanyal, B. K. Mishra and P. M. Puri, J. Phys. Chem. B, 2001, 105, 12823–12831 CrossRef CAS; (b) I. M. Umlong and K. Ismail, J. Colloid Interface Sci., 2005, 291, 529–536 CrossRef CAS; (c) Y. Fan, Y. Li, G. Yuan, Y. Wang, J. Wang, C. C. Han, H. Yan, Z. Li and R. K. Thomas, Langmuir, 2005, 21, 3814–3820 CrossRef CAS.
- O. Ghosh and C. A. Miller, J. Phys. Chem. B, 1987, 91, 4528–4535 Search PubMed.
- I. Grillo, E. I. Kats and A. R. Muratov, Langmuir, 2003, 19, 4573–4581 CrossRef CAS.
- J. I. Briz and M. M. Velázquez, J. Colloid Interface Sci., 2002, 247, 437–446 CrossRef CAS.
- N. Gospodinova, P. Mokreva and L. Terlemezyan, Polymer, 1993, 34, 2438–2439 CrossRef CAS.
- Y.-G. Han, T. Kusunose and T. Sekino, Synth. Met., 2009, 159, 123–131 CrossRef CAS.
- (a) P. N. Adams, P. J. Laughlin, A. P. Monkman and A. M. Kenwright, Polymer, 1996, 37, 3411–3417 CrossRef CAS; (b) J. Stejskal, A. Riede, D. Hlavatá, J. Prokeš, M. Helmstedt and P. Holler, Synth. Met., 1998, 96, 55–61 CrossRef CAS.
- For the estimation of the maximal number of bound HRP molecules, the following assumptions were made: (1) average vesicle diameter of 100 nm; (2) unilamellarity of the vesicles; and (3) AOT head group area of 67 Å2: I. Grillo, P. Levitz and Th. Zemb, Langmuir, 2000, 16, 4830–4839 Search PubMed.
- S. K. Manohar and A. G. MacDiarmid, Synth. Met., 1991, 41–43, 711–714 CrossRef.
- M. T. Gill, S. E. Chapman, C. L. DeArmitt, F. L. Baines, C. M. Dasdswell, J. G. Stamper, G. A. Lawless, N. C. Billingham and S. P. Armes, Synth. Met., 1998, 93, 227–233 CrossRef CAS.
- M. Radoičić, Z. Šaponjić, J. Nedeliković, G. Ćirić-Marjanović and J. Stejskal, Synth. Met., 2010, 160, 1325–1334 CrossRef CAS.
- A. J. Milton and A. P. Monkman, J. Phys. D: Appl. Phys., 1993, 26, 1468–1474 CrossRef CAS.
- M. Aizawa, L. Wang, H. Shinohara and Y. Ikariyama, J. Biotechnol., 1990, 14, 301–310 CrossRef CAS.
- F. Wudl, R. O. Angus, Jr, F. L. Lu, P. M. Allman, D. J. Vachon, M. Nowak, Z. X. Liu and A. J. Heeger, J. Am. Chem. Soc., 1987, 109, 3677–3684 CrossRef CAS.
- (a) S. M. Marinakos, M. F. Anderson, J. A. Ryan, L. D. Martin and D. L. Feldheim, J. Phys. Chem. B, 2001, 105, 8872–8876 CrossRef CAS; (b) X. Feng, C. Mao, G. Yang, W. Hou and J.-J. Zhu, Langmuir, 2006, 22, 4384–4389 CrossRef CAS; (c) Y. Zhu, D. Hu, M. Wan, L. Jiang and Y. Wei, Adv. Mater., 2007, 19, 2092–2096 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |