Directed co-assembly of heme proteins with amphiphilic block copolymers toward functional biomolecular materials
Andrew D.
Presley
a,
Joseph J.
Chang
a and
Ting
Xu
*abc
aDepartment of Materials Science and Engineering, University of California, Berkeley, CA 94720, USA
bDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: tingxu@berkeley.edu
cMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 21st October 2010
Abstract
Directed co-assembly of block copolymers and proteins/peptides may lead to hierarchically structured functional biomolecular materials. However, this requires one to synergistically direct multiple self-assembly processes. Retaining proper cofactor binding is essential to utilize many bio-motifs for catalytic reactions and sensing. Here, by using a heme-binding helix bundle peptide-polymer conjugate and a holomyoglobin-polymer conjugate as examples, we show that the simultaneous, macroscopic assembly of heme-binding proteins and diblock copolymers can be achieved in thin films without compromising protein structures, cofactor binding and enzymatic activities. To our knowledge, this is the first example of a protein/cofactor complex formed upon being co-assembled with an amphiphilic block copolymer in thin films. Molecular assemblyvia a combination of biomolecular recognition and polymer phase separation in this fashion will lead to hybrid materials combining properties of both synthetic and biological building blocks.
Introduction
Hybrid materials based on synthetic polymers and natural building blocks, such as peptides and proteins, have the potential to combine the advantages of both building blocks and lead to functional assemblies spanning multiple length scales.1,2 Integration of biomolecules with amphiphilic block copolymers (BCPs) is particularly interesting. BCPs microphase separate into well defined microdomains that are typically tens of nanometers in size.3Protein incorporation could endow BCPs with molecularly-defined hierarchical structures and important new functionalities, such as catalysis, transport, chemical sensing, and signaling with superior sensitivity and selectivity. The lateral ordering and macroscopic orientation of BCP microdomains in thin films can be readily tailored4–6 to translate the molecular-level structural elements and diverse functionalities of proteins across the nanometer- and micron-regimes to macroscopic length scales.The generation of functional hybrid biomolecular materials relies on the synergistic integration of two self-assembly processes, namely protein folding and BCP phase separation. Co-assembly must be conducted so as to retain BCP phase separation as well as protein structure and functionality. Additionally, the unique functionalities of many proteins hinge on non-covalent binding of small-molecule cofactors.7,8 Therefore processing conditions for hybrid materials must also be chosen to preserve these cofactor complexes or to promote their reconstitution. Simultaneous manipulation of these processes represents a significant challenge. To date, nanoscopic protein patterning using BCP thin films has been achieved primarily through a stepwise process in which the films are first fabricated independently and then subjected to protein adsorption from aqueous solution.9–15 Such assemblies have been utilized as platforms for immunoassays and to tailor cell adhesion and proliferation.12,16–19
A simultaneous co-assembly strategy for BCP/protein hybrids is desirable as the resulting composites may exhibit hierarchical structural complexity that is integral to the material rather than appended as a discrete surface layer. Furthermore, a co-assembly strategy may enable enhanced processibility for hybrid materials containing natural proteins. Few studies have investigated the simultaneous co-assembly of BCPs and proteins in this manner. Some have utilized Pluronics and a limited number of other copolymers for direct incorporation of proteins such as hemoglobin in BCP thin films.20,21 A notable example demonstrated incorporation of ferritin-based nanoparticles in a poly(2-vinylpyridine)-b-poly(ethylene oxide) (P2VP-b-PEO) thin film.22 The resulting composite contained ferritin-PEO nanoparticles confined to PEO microdomains, showing that direction of protein localization with BCP films is possible. However it is not yet clear that proteins lacking ferritin's exceptional stability can be used as building blocks in BCP-based hybrid biomaterials. Furthermore, generation of nanostructured thin films viaco-assembly of amphiphilic BCPs with catalytically active enzymes has not been demonstrated to our knowledge. Preservation or reconstitution of functional cofactor complexes in these cases may be required during the co-assembly process, although this has yet to be established experimentally.
Two strategies are used here to investigate the proposed co-assembly process. One employs de novo designed protein tertiary motifs, coiled-coil helix bundles, that have simple and robust structures and mimic certain functionalities of natural proteins.23,24 Helix bundles have been designed for catalytic oxidation of small phenolic substrates25 as well as for light-induced charge separation and electron transfer for artificial photosynthesis.26,27 These functional motifs are based on peptides with relatively short sequences, typically 20–40 residues in length, and have shown exceptional stability against temperature, pH and organic solvents in comparison to their natural counterparts.28,29 Interior binding pockets can be engineered to bind small-molecule cofactors with high selectivity and sensitivity,8,30 requisite for materials applications such as catalysis or chemical sensing. On the other hand, many naturally occurring proteins and enzymes exhibit sophisticated functionalities that are unmatched by designed analogues. So, a second strategy employs more complex proteins that are conjugated with synthetic polymers to provide compatibility with the BCP component and to enhance stability during the assembly process. In either strategy, directing the co-assembly of active protein/cofactor complexes with amphiphilic BCPs may lead to high density arrays of these functional elements to achieve functional biomolecular materials.
Here we report the simultaneous co-assembly of an amphiphilic BCP, polystyrene-b-poly(ethylene oxide) (PS-b-PEO), with a model heme-binding protein motif, the coiled-coil α-helix bundle, and a natural heme protein, horse-heart myoglobin (Mb), to form hierarchically structured films as shown schematically in Fig. 1. Through a synergistic co-assembly process, protein structure was retained, heme-binding by helix bundles was reconstituted, and retention of Mb peroxidase activity was observed. Hierarchical organization of these multiple functional elements through a combination of protein folding, biomolecular recognition and BCP microphase separation represents a new approach to fabricate functional biomolecular materials using peptides and enzymatically active proteins. This work demonstrates the synergistic co-assembly of BCPs, peptides/proteins, and small-molecule cofactors, providing insight for further development of this new family of hybrid materials.
 | ||
| Fig. 1 Hierarchical assembly of peptide-polymer/block-copolymer (BCP) thin films. Multi-component samples are blended in solution and processed into thin films. Macroscopic BCP thin films contain phase-separated nanoscale domains (shown in blue and brown). Helix bundle peptides (shown in green), displaying characteristic angstrom-scale secondary structure, are sequestered within the BCP domains. Synthetic polymers are conjugated to the bundle periphery in order to mediate interactions with the BCP. Small molecule cofactors (shown in red) are bound with molecular-level precision by the peptide bundles. Here we depict the assembly of heme-containing four-helix bundles to emphasize the reconstitution of biological complexes during film processing. We have extended the approach to include horse-heart myoglobin (Mb), a naturally-occurring heme protein. | ||
Experimental
Synthesis of peptide-PEO conjugates
The heme-binding coiled-coil 4-helix bundle peptide, termed H10H24 (AcNH-GGGEIWKLHEEFLCKFEELLKLHEERLKKM-COOH), was synthesized using Fmoc solid-phase peptide synthesis as previously reported.31 Briefly, peptides were synthesized on a 50-μmole scale on Wang resin. N-termini were acetylated with acetic anhydride. Global deprotection of amino acid sidechains was conducted concurrently with cleavage from the solid support. Maleimide-functionalized PEO (Mw = 2000 g/mol, Rapp Polymere, Germany) was conjugated to the peptidesvia reaction with cysteine (C14) sidechain thiols. Modification of crude H10H24 was typically conducted with a five-fold excess of maleimide-PEO in 25 mM potassium phosphate buffer, pH = 8, for 30–60 min at room temperature. Synthesis of peptides and peptide-polymer conjugates was monitored using analytical reverse-phase high performance liquid chromatography (HPLC) and MALDI-TOF mass spectrometry, and the desired conjugates were purified via preparative-scale HPLC.Synthesis of myoglobin-PEO conjugate
Horse heart myoglobin (Mb, Sigma) was dissolved in 25 mM phosphate buffer, pH = 7.2, to a concentration of 1 mM and reacted with 2-iminothiolane (2-IT, 30 eq) for 1 h at room temperature. Excess 2-IT was removed by buffer exchange into 25 mM phosphate, pH = 8, containing 1 mM TCEP using a NAP-5 desalting column (GE Healthcare). Maleimide-terminated PEO (10 eq, Mw = 2000 g/mol) was added to the Mb solution and the reaction mixture was incubated overnight at room temperature. Buffer salts were removed by exchange into deionized H2O using a NAP-10 desalting column, and excess polymer was removed by repeated dilution in H2O and concentration using an AmiconUltra spin concentrator (MWCO = 10,000 Da; Millipore). The resulting samples were free of excess polymer as judged by HPLC and Mb concentration was in excess of 50 mg/mL. SDS-polyacrylamide gel electrophoresis was conducted according to the method of Laemmli.32 Following electrophoresis, protein bands were visualized by Coomassie staining.Preparation of BCP/peptide blends
PS(19,000)-b-PEO(6,400) (PDI = 1.08) was purchased from Polymer Source, Inc. (Montreal, Quebec, Canada). A 1.1% (w/v) solution of PS-b-PEO was prepared in benzene, and peptide solutions were prepared in methanol. H10H24-P2K solutions contained 0.9–6.8% (w/v) peptide-conjugate (1.6–12 mM) in methanol. Solutions were quantified by UV absorbance of tryptophan as described below. Porcine hemin (1.9–14 mM, Fluka) was prepared in benzene containing 20% (v/v) DMSO. The PS-b-PEO solution (270 μL) was diluted with methanol or a methanol/benzene mixture (80 μL), followed by addition of the peptide solution (30 μL) and heme solution (20 μL).Preparation of BCP/Mb blends
Protein solutions containing 0.03–2.0% Mb-PEO (∼12–800 μM) were prepared using 80% (v/v) methanol in water. The resulting Mb-PEO solutions (30 μL) were diluted with additional methanol (80 μL), followed by PS-b-PEO solution (1.1% in benzene, 270 μL).Fabrication of thin films
Silicon substrates were cleaned for two minutes with air plasma in a PDC-001 Plasma Cleaner (Harrick). 1′′ polished quartz plates (Quartz Scientific, Inc.) were cleaned in Nochromix detergent (Godax) and rinsed thoroughly with copious DI water prior to plasma cleaning. Thin films on silicon and quartz were prepared by spin-coating the PS-b-PEO/peptide blends for 10 s, with varying speed for control of film thickness. Helix bundle films contained 6–40% (w/w) H10H24 or H10H24-P2K and 0.8 molar equivalents of heme per peptide, and film thickness was 50–200 nm as measured using a Filmetrics F20 interferometer. Mb films contained ∼0.3–20% (w/w) Mb-PEO, and ranged in thickness between 50–100nm.Samples were solvent-annealed using a mixture of water and toluene. The samples were placed in 2.6-L cylindrical aluminium chamber equipped with an O-ring-sealed lid. Samples were annealed in the chamber containing DI water (6 mL) for 2 h; benzene (6 mL) was added to the chamber and annealing was continued for 4–16 h. The temperature was maintained at 35 °C with a fiberglass-insulated heater tape, controlled by a CN7523 temperature controller in conjunction with a 25-amp solid state relay and a J-type surface thermocouple (Omega).
To prevent delamination of the films from the substrates during enzymatic assays in aqueous solution, silicon wafers were modified with a hydrophobic brush consisting of poly(styrene-r-benzocyclobutene), called P(S-r-BCB). The P(S-r-BCB) (MW = 90,000 g/mol; 8% BCB; PDI = 1.4) was the kind gift of T.P. Russell, and substrates were prepared according to a previously published procedure.33
Mb peroxidase activity assay
Following thin film preparation, Mb peroxidase activity was assayed by placing a thin film in 1 mL of TMB peroxidase assay reagent (TMB Peroxidase EIA Substrate Kit #172–1067, Bio-Rad) prepared according to the manufacturer's instructions. The enzymatic reaction was allowed to proceed at room temperature for 5–20 min.Atomic force microscopy (AFM)
AFM was performed on a Molecular Imaging PicoSPM II with a PicoScan 2500 controller using doped silicon cantilevers (TESP model, Veeco, Inc). Cantilever spring constants were 20–80 N/m with resonant frequencies of 296–341 kHz. Images were acquired using a scan rate of 2 Hz.Circular dichroism (CD)
CD was performed on a Jasco J810 spectropolarimeter. Samples prepared as above were cast and annealed on quartz plates, and spectra were recorded at ambient temperature between 190 and 260 nm at 1-nm intervals using a 1-nm bandwidth and a 4-second response time.UV-visible absorption spectroscopy (UV-Vis)
UV-vis spectra were recorded on a Hewlett-Packard 8453 spectrophotometer. Solution samples were analyzed using standard 1-cm path length cuvettes, and concentrations were determined by tryptophan absorbance at 280 nm assuming an extinction coefficient of 5500 L mol−1 cm−1. Thin films were analyzed following casting and annealing of PS-b-PEO/protein blends on quartz plates as described above.Grazing-incidence small angel X-ray scattering (GISAXS)
GISAXS measurements were made on beamline 7.3.3 at the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory (LBNL) and beamline 8-ID-E at the Advanced Photon Source at Argonne National Laboratory (APS-ANL). Scattering profiles were collected on an ADSC Quantum 4u CCD detector at ALS and a Mar-CCD detector at APS. Line-averaged intensities are reported as I vs. q, where q = (4π/λ)sin(θ/2), λ is the wavelength of the incident X-rays, and θ is the scattering angle. X-ray reflectivity profiles were recorded at APS.Results and discussion
PS-b-PEO was used as a model BCP because it contains a protein-compatible PEO block and the PEO microdomains may provide a hydrated environment conducive to proper protein folding and function. Most proteins are temperature sensitive and unfold upon exposure to elevated temperatures. PS-b-PEO readily ordered in thin films viasolvent annealing forming hexagonally-packed PEO cylinders oriented normal to the surface.34,35Solvent annealing was conducted at a moderate temperature (35 °C), which may allow for prevention of protein denaturation and retention of secondary and tertiary structure. The coiled-coil α-helix bundle was chosen because it is one of the simplest tertiary peptide motifs that retains a specific function of interest here, namely small-molecule cofactor binding.8,23,26,36,37 The heme-binding coiled-coil 4-helix bundle used here, termed H10H24, was initially designed as a model protein motif for the study of more complex heme-binding proteins.26,36,38–41 It has simple and robust secondary and tertiary structures and presents an ideal starting point to explore the principles governing the co-assembly of natural proteins with synthetic BCPs.8 A naturally occurring heme protein, horse-heart myoglobin (Mb), was also used to assess the structure and function of more complex proteins within BCP thin films.Co-assembly of amphiphilic BCPs and biological building blocks is a challenge, limited by the chemical incompatibility of the two classes of materials. Peptides and proteins naturally exist in aqueous solution at or near physiological pH and usually have poor solubility in organic solvents.42,43 Amphiphilic BCPs, on the other hand, are frequently processed using volatile hydrocarbon solvents which can denature proteins and peptides. Recent developments in polymer-protein conjugation may be used to enhance compatibility and overcome some of these hurdles.44–48 Simultaneous assembly, however, requires co-solubilization of the BCP and protein which constitutes a significant obstacle. Conditions must be chosen so as to maintain peptide structure, as well as preserve binding of necessary cofactors in solution or promote reconstitution of the cofactor complexes during film processing. Meanwhile, the system must allow for ready fabrication of the BCP thin films. Solvents should be sufficiently volatile to allow for film casting, as well as subsequent annealing if ordered materials are desired.
Before protein-containing thin films could be cast, it was therefore necessary to find a solvent system that provided adequate solubility for all of the component materials. PS-b-PEO was dissolved in benzene, a good solvent for both blocks of the copolymer. However, H10H24 and Mb are insoluble and precipitated in neat benzene as well as many other solvents suitable for thin film processing. Several co-solvents ranging in polarity were tested to solubilize blends of PS-b-PEO and heme-binding proteins/peptides. Peptide solubility improved when polar co-solvents were added. Methanol, although a non-solvent for PS, was found to best solubilize the blends of PS-b-PEO and H10H24 while still allowing for casting of thin films with uniform thickness. However, Mb could not be solubilized using co-solvents. To improve the solubility and stability of the proteins in organic solvents, PEO was conjugated to protein side chains. PEO conjugation has been shown to improve protein solubility in organic solvents and stabilize protein structures against extremes in temperature, pH, and ionic strength.31,49–51Maleimide-terminated PEO (2000 g/mol) was attached to C14 on the bundle periphery of H10H24 as described previously to form a peptide-polymer conjugate, termed H10H24-P2K.31 Native Mb was also covalently modified with PEO to impart solubility in organic solvents as discussed in detail below.
After methanol was found to be a suitable cosolvent, H10H24 and H10H24-P2K were first dissolved in methanol and combined with a stock solution of 1% PS-b-PEO (w/v) in benzene. A series of samples containing 2–40% (w/w) peptide and 10–33% (v/v) methanol were prepared. 33% methanol by volume was required to maintain peptide solubility at weight fractions above 20%. Heme (i.e.chloroprotoporphyrin IX iron (III)) was dissolved in dimethylsulfoxide (DMSO), diluted with benzene or methanol, and added to the peptide/BCP blends. The samples contained 0.8 equivalents of heme per peptide, and the final DMSO content for all heme-containing samples was ≤5% by volume. Films with thicknesses ranging from 50 to 200 nm were prepared by spin-casting the solutions of peptide/BCP blends onto silicon substrates for structural analysis or quartz substrates for spectroscopic analysis. The films were solvent-annealed at 35 °C in a chamber saturated with water vapor for two hours followed by water/benzene vapor for four hours.
Addition of H10H24 or H10H24-P2K did not interfere with the microphase separation of PS-b-PEO or the vertical alignment of PEO microdomains and lateral ordering in the thin films. AFM images for a solvent-annealed, 60-nm film containing 12% (w/w) H10H24-P2K in PS-b-PEO are shown in Fig. 2. Similar AFM images were seen for blends of unmodified H10H24 and PS-b-PEO. Hexagonally packed cylindrical microdomains oriented normal to the surface can be clearly seen. A range of peptide loadings was investigated. H10H24-P2K weight fractions were varied using an upper limit of 40% (w/w). When cast from benzene solution containing 33% (v/v) methanol, films with less than 20% (w/w) H10H24-P2K exhibited large defects. These defects were minimized by reducing the methanol content of the benzene solution to 10% (v/v) (see Supporting Information, Figure S1). The micrographs in Fig. 2 are typical of most of the films, exhibiting order similar to films of PS-b-PEO alone.
 | ||
| Fig. 2 Characterization of peptide-conjugate/BCP thin films by atomic force microscopy (AFM). AFM a) height image and b) phase image of a PS-b-PEO thin film containing 12% (w/w) H10H24-P2K conjugate and 0.8 eq heme per peptide. Scale bar = 400 nm. The film was annealed at 35 °C for 2 h in a water atmosphere, followed by 4 h in a water/benzene atmosphere. | ||
Maintenance of protein structure and function within the nano-structured thin films is of primary importance. Peptide secondary structure was assessed via CD. CD data for a PS-b-PEO film containing holo-form 40% (w/w) H10H24-P2K with 0.8 equivalents of heme per peptide are shown in Fig. 3 (blue circles). The CD spectrum exhibits local minima at 208–210 nm and 220–223 nm, characteristic of α-helices, with a ratio of ∼1. The CD studies indicate that helical character of peptide is maintained when the helix bundle-PEO conjugates are co-assembled with PS-b-PEO in thin films. Similar features were observed for thin films containing 12–40% (w/w) H10H24-P2K before and after solvent annealing, with and without heme inclusion. CD signal could not be detected for films with lower peptide contents, due to limited instrument sensitivity. As observed in solution experiments,31PEO conjugation generally lead to enhanced helicity as judged by ellipticity at 222 nm and H10H24-P2K consistently exhibited greater ellipticity than the unmodified helix bundle. This is evident when comparing CD spectra for films containing H10H24-P2K and an equimolar amount of unmodified H10H24 in Fig. 3. The raw CD data used in this comparison are not normalized for peptide concentration, because the peptide in the thin film is not present in levels high enough to quantity by UV-vis absorption spectroscopy. Peptide concentrations in solution and the film thickness were equal across samples, however, so we attribute the greater ellipticity of H10H24-P2K to helix stabilization stemming from PEO conjugation.
 | ||
| Fig. 3 CD analysis of PS-b-PEO/peptide thin films. a) CD spectra of annealed thin films containing equimolar amounts of H10H24 (24% (w/w), red circles) and H10H24-P2K (40% (w/w), blue circles). Raw data are plotted as individual points; spectra refined via binomial smoothing are plotted in grey. The H10H24-P2K conjugate exhibits greater helicity than unmodified H10H24. | ||
The oligomeric state of coiled-coil helix bundles (i.e.peptide tertiary structure) in solution is typically assessed via analytical ultracentrifugation, but this technique is not applicable to the present thin film system. A qualitative measure of tertiary structure can be made by examining the core functionality of the peptide bundle (namely, cofactor binding). Binding of heme and other porphyrin-based cofactors is readily evaluated by UV-visible absorption spectroscopy, as binding leads to distinctive spectroscopic features that have been studied in detail.30,38,41
UV-vis absorption spectra were recorded for a series of PS-b-PEO samples containing 40% (w/w) H10H24-P2K and 0.8 equivalents of heme per peptide (Fig. 4a). Prior to spin-casting of thin films, the heme Soret band in benzene/methanol solution is observed at 398 nm (Fig. 4a, black trace). This spectrum is characteristic of free heme in solution, as is to be expected in this nonpolar solvent system.52Spin-casting of the thin films is accompanied by a slight red-shifting of the Soret maximum to 403 nm in the corresponding UV-vis spectrum (Fig. 4a, red trace). Most significantly, further red shift of the Soret maximum to 413 nm and resolution of the porphyrin Q-bands are observed upon solvent annealing (Fig. 4a, blue trace). This is consistent with heme binding by H10H24viabis-histidyl ligation.38 This is not observed for films containing peptides that do not bind heme (Fig. 4b, black trace) or for films that lack peptide altogether (Fig. 4b, red trace). On the contrary, those samples demonstrate broadening and blue-shifting of the spectral features suggesting π–π aggregation of unbound heme.53,54 Taken together the UV-vis data confirm that the H10H24 bundles are properly assembled for heme-binding, which is consistent with the preservation of helix bundle tertiary structure within the phase-separated PS-b-PEO thin film. These results suggest that the heme is unbound in organic solution and remains so in as-cast films. The annealing process provides sufficient mobility to allow sequestration of heme within the helix-bundles viabis-histidyl ligation. This significant result demonstrates the feasibility of organizing multiple functional building blocks in a specific manner via naturally occurring and engineered biomolecular host–guest interactions in multi-component systems.
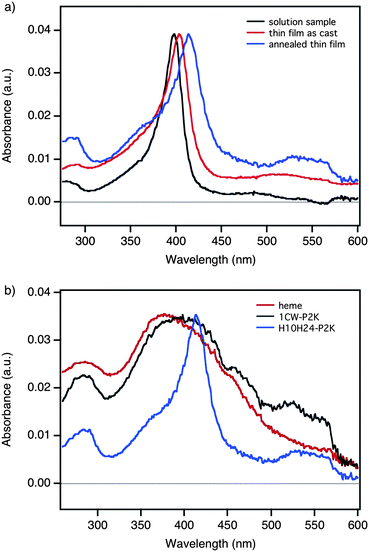 | ||
| Fig. 4 Analysis of heme binding by H10H24-P2K in PS-b-PEO thin films. a) UV-vis spectra for films containing 40% (w/w) H10H24-P2K and 0.8 equivalents of heme: in benzene/methanol solution (black); as cast in films (red); and in annealed films (blue). Red-shifting of the Soret band to 414 nm upon annealing is indicative of bis-histidyl ligation of heme by H10H24-P2K bundles. b) UV-vis spectra for films containing heme and: 40% (w/w) H10H24-P2K (blue); 40% (w/w) 1CW-P2K (black); and no peptide (red). Thin films lacking H10H24-P2K exhibit broad, blue-shifted features due to heme aggregation. | ||
Helix bundles including H10H24 were de novo designed as simplified models for more complex heme-binding proteins. Their utility arises primarily from their structural simplicity and robust, reversible folding processes. We also investigated PS-b-PEO thin films containing Mb in order to assess the feasibility of including more sensitive, naturally-occurring proteins in BCP blends. Conversion of Mb lysine residues to thiols using 2-iminothiolane and subsequent reaction with maleimide-functionalized PEO led to an Mb-PEO conjugate containing multiple polymer chains per peptide (Mw ∼30 kDa) as observed by SDS-PAGE (Fig. 5a-b). The Mb-PEO conjugate was readily blended with PS-b-PEO in benzene/methanol solution, whereas the native protein precipitated from blended samples (Fig. 5c). Before casting the blended samples, silicon substrates were modified with a crosslinked polystyrene brush to prevent film dewetting during enzymatic assays in aqueous solution.33 As observed for H10H24-P2K, incorporation of the Mb-PEO conjugate did not interfere with BCP microphase separation, and hexagonally-packed cylinders were observed for the Mb-PEO/PS-b-PEO thin films with similar solvent annealing (Fig. 5d).
 | ||
| Fig. 5 Preparation and analysis of Mb-PEO conjugates and Mb-PEO/PS-b-PEO thin films. a) The Mb-PEO conjugate is synthesized via conversion of Mb lysine residues to thiols using 2-iminothiolane and subsequent reaction with maleimide-terminated PEO (Mw = 2000 Da). b) SDS-PAGE analysis indicates covalent attachment of several PEO chains per Mb molecule. c) Native Mb precipitates from PS-b-PEO solution samples, while Mb-PEO remains soluble. d) AFM height image of a PS-b-PEO thin film containing 17% (w/w) Mb-PEO (scale bar = 400 nm). Hexagonally packed PEO cylinders with good lateral ordering are observed. e) Mb peroxidase activity is maintained in PS-b-PEO thin films. Mb-containing films oxidize TMB in the presence of hydrogen peroxide to form blue reaction product. The photograph was taken after 15 min of reaction time at room temperature. | ||
To assess protein integrity, the thin films were assayed for Mb peroxidase activity. Oxidation of tetramethylbenzidene (TMB) in the presence of hydrogen peroxide was observed for PS-b-PEO films containing the Mb-PEO conjugate (Fig. 5e), qualitatively indicating that the protein remained catalytically active. Control experiments assaying apo-Mb and unbound heme in solution resulted in negligible activity as assessed by the TMB analysis (data not shown). A thin film containing 17% (w/w) Mb-PEO with an area of 1.6 cm2 and an average thickness of 70 nm was estimated to contain 1.2 μg of Mb, assuming a film density similar to the density of the PS-b-PEO (∼1 g/mL). When activity for this thin film sample was quantified against standard solution samples containing known amounts of unmodified Mb, the measured activity was found to be ∼40% higher than the expected value, equivalent to 1.7 μg of Mb. Due to the possible error in the estimated Mb content, this observed enhancement still needs further verification. Nevertheless, it is clear that the Mb-PEO conjugate can be incorporated in the thin films with preservation of protein structure and activity.
AFM results indicate that the protein-conjugate/PS-b-PEO thin films self-assemble into nanoscale morphologies similar to the thin films of PS-b-PEO alone. CD, heme binding experiments, and the TMB peroxidase assay confirmed the integrity of the H10H24-P2K bundles and Mb-PEO. However, the spatial distribution of the protein-PEO conjugates within the thin film, and whether they are indeed sequestered in the PEO cylinders, remains a question. Based on AFM micrographs, we conclude that H10H24-P2K and Mb-PEO do not phase separate to the film surface. X-ray reflectivity profiles are consistent with films consisting of a single layer, indicating that conjugates also do not separate at the PS-b-PEO/silicon oxide interface (see Supporting Information, Figure S2).
GISAXS was used to characterize macroscopic morphology and ordering of co-assembled PS-b-PEO and H10H24-P2K. To ensure consistency in the solvent annealing conditions, thin films of blends with different protein-PEO conjugate contents were annealed in the same chamber with the same treatment. Fig. 6a shows the GISAXS pattern for a thin film containing 13% (w/w) H10H24-P2K at an incident angle of 0.18°. The corresponding qy scan at qz = 0.02 Å−1 is shown in Fig. 6b. The films exhibit diffraction peaks with relative positions at q, √3q, √4q, and √7q as expected for hexagonally packed PEO cylinders oriented normal to the substrate surface. The first-order scattering peak for PS-b-PEO films without peptide occurs at qy = 0.017 Å−1, corresponding to a periodicity of 36.4 ± 0.4 nm. In thin films of the H10H24-P2K blend, cylinder periodicity is 33.1 ± 0.5 nm (qy = 0.019 Å−1), constituting a 9% decrease. In a control experiment, films containing a stoichiometrically equivalent amount of PEO (2000 g/mol) exhibited an expected increase in cylinder spacing (see Supporting Information, Figure S3). This indicates that peptide secondary and tertiary structure has a substantial impact on the segregation behavior of the peptide-polymer conjugate within the thin film. While absolute periodicity changes varied from one sample set to another, comparisons between films with and without H10H24-P2K were made for samples that were annealed together in the same chamber under identical conditions. It should be noted that the X-ray beam footprint in the GISAXS geometry is approximately 1.5 cm, so the observed GISAXS patterns reflect ordering of the blends over macroscopic distances. Inter-cylinder spacing was consistently found to decrease upon blending with H10H24 and H10H24-P2K, and similar results were observed for the blends of PS-b-PEO and Mb-PEO (see Supporting Information, Figure S4). This periodicity reduction confirmed the co-assembly of protein-PEG conjugates within the PS-b-PEO thin films. We speculate that the periodicity reduction may be due to differences in solvent selectivity for proteinvs. the PEO homopolymer during the annealing process, as well as possible differences in chain packing upon inclusion of protein-PEO conjugates within the BCP microdomains. We are currently carrying out resonant soft X-ray scattering studies to identify the exact location of peptide/protein-PEO conjugates in the thin films.
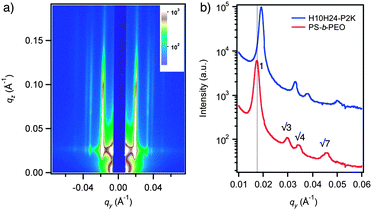 | ||
| Fig. 6 GISAXS analysis of peptide/BCP thin films. a) GISAXS scattering pattern for PS-b-PEO thin film at incident angle α = 0.15°. The pattern is characteristic of hexagonally-packed cylinders oriented normal to the substrate surface. b) qy intensity profiles, taken at qz = 0.02, for PS-b-PEO films containing no peptide (red) and 12% (w/w) H10H24-P2K (blue). Peptide incorporation generally leads to a decrease in PEO cylinder spacing. | ||
Conclusions
In conclusion, we have demonstrated the co-assembly of heme binding peptide/protein-PEO conjugates in block copolymer thin films. Solubility limitations were overcome through the use of a co-solvent system that allowed for simultaneous processing of copolymer, peptides, and heme cofactors. Modification of proteins with PEO provided compatibility with the copolymer component in organic solution and allowed for film casting. However, polymer conjugation is not required for simpler protein functional motifs. The thin films exhibited macroscale lateral ordering and regular nanoscale morphologies that are characteristic of the BCP component. Secondary and tertiary structure of the helix bundle were maintained as assessed by circular dichroism and peptide-dependent heme binding. To our knowledge, this is the first demonstration of the formation of a protein/cofactor complex within a BCP thin film. In this manner we expect to endow BCP nanostructures with molecularly-defined structural and functional elements that cannot be realized by the polymer alone, thereby creating new hierarchically ordered materials with morphological control on multiple length scales. Because coiled-coil helix bundles and other de novo designed peptide structures can be engineered for specific recognition and binding of various natural and non-natural cofactors,8,30,55co-assembly of BCPs with these designed structures may therefore lead to novel materials possessing the biocatalytic characteristics of enzymes or altogether new chemical and physical properties.41,55 Our model system was extended to include a heme-containing enzyme, horse-heart myoglobin, demonstrating that catalytically active enzymes can also be co-assembled in the BCP thin films. We have observed activity losses upon storage of the Mb-PEO films as well as leaching of the protein from the films upon exposure to aqueous assay media, so we are currently conducting a thorough study of enzyme lifetime and exploring alternative methods for enzyme immobilization within the thin films. Upon further engineering and application to other enzymes, this co-assembly approach could potentially allow for fabrication of thin films with a range of desirable functionality. The studies reported here will guide further co-assembly of natural proteins with synthetic polymers to achieve functional hybrid materials.Acknowledgements
This work is supported by the U.S. Department of Energy through the Hybrid Biomaterials Scattering Program at Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH11231. Use of the Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We acknowledge helpful discussions with Jessica Y. Shu and Yu-Ja Huang regarding spectroscopic characterization of peptide/BCP thin films.References
- H.a. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- G. W. M. Vandermeulen and H. A. Klok, Macromol. Biosci., 2004, 4, 383–398 CrossRef CAS.
- M. J. Fasolka and A. M. Mayes, Annu. Rev. Mater. Res., 2001, 31, 323–355 CrossRef CAS.
- S. H. Kim, M. J. Misner and T. P. Russell, Adv. Mater., 2004, 16, 2119 CrossRef CAS.
- S. Park, D. H. Lee, J. Xu, B. Kim, S. W. Hong, U. Jeong, T. Xu and T. P. Russell, Science, 2009, 323, 1030 CrossRef CAS.
- T. Xu, J. T. Goldbach and T. P. Russell, Macromolecules, 2003, 36, 7296–7300 CrossRef CAS.
- M. H. Hecht, A. Das, A. Go, L. H. Bradley and Y. N. Wei, Protein Sci., 2004, 13, 1711–1723 CrossRef CAS.
- R. L. Koder and P. L. Dutton, Dalton Trans., 2006, 3045–3051 RSC.
- N. Kumar and J. I. Hahm, Langmuir, 2005, 21, 6652–6655 CrossRef CAS.
- N. Kumar, O. Parajuli, A. Dorfman, D. Kipp and J. I. Hahm, Langmuir, 2007, 23, 7416–7422 CrossRef CAS.
- P. A. George, K. Quinn and J. J. Cooper-White, Biomaterials, 2010, 31, 641–647 CrossRef CAS.
- K. H. A. Lau, J. Bang, D. H. Kim and W. Knoll, Adv. Funct. Mater., 2008, 18, 3148–3157 CrossRef CAS.
- Q. Li, K. H. A. Lau, E. K. Sinner, D. H. Kim and W. Knoll, Langmuir, 2009, 25, 12144–12150 CrossRef CAS.
- D. Liu, T. Wang and J. L. Keddie, Langmuir, 2009, 25, 4526–4534 CrossRef CAS.
- M. Matsusaki, M. Omichi, K. Kadowaki, B. H. Kim, S. O. Kim, I. Maruyama and M. Akashi, Chem. Commun., 2010, 46, 1911–1913 RSC.
- H. L. Khor, Y. Kuan, H. Kukula, K. Tamada, W. Knoll, M. Moeller and D. W. Hutmacher, Biomacromolecules, 2007, 8, 1530–1540 CrossRef CAS.
- P. A. George, M. R. Doran, T. I. Croll, T. P. Munro and J. J. Cooper-White, Biomaterials, 2009, 30, 4732–4737 CrossRef CAS.
- J. H. Seo, R. Matsuno, M. Takai and K. Ishihara, Biomaterials, 2009, 30, 5330–5340 CrossRef CAS.
- C. M. Grozea, N. Gunari, J. A. Finlay, D. Grozea, M. E. Callow, J. A. Callow, Z. H. Lu and G. C. Walker, Biomacromolecules, 2009, 10, 1004–1012 CrossRef CAS.
- W. J. Shan, P. L. He and N. F. Hu, Electrochim. Acta, 2005, 51, 432–440 CrossRef CAS.
- S. S. Jia, J. J. Fei, J. J. Deng, Y. L. Cai and J. A. Li, Sens. Actuators, B, 2009, 138, 244–250 CrossRef.
- Y. Lin, A. Boker, J. B. He, K. Sill, H. Q. Xiang, C. Abetz, X. F. Li, J. Wang, T. Emrick, S. Long, Q. Wang, A. Balazs and T. P. Russell, Nature, 2005, 434, 55–59 CrossRef CAS.
- W. F. DeGrado, C. M. Summa, V. Pavone, F. Nastri and A. Lombardi, Annu. Rev. Biochem., 1999, 68, 779–819 CrossRef CAS.
- T. Xu and J. Shu, Soft Matter, 2010, 6, 212–217 RSC.
- O. Maglio, F. Nastri, V. Pavone, A. Lombardi and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3772–3777 CrossRef CAS.
- S. X. Ye, B. M. Discher, J. Strzalka, T. Xu, S. P. Wu, D. Noy, I. Kuzmenko, T. Gog, M. J. Therien, P. L. Dutton and J. K. Blasie, Nano Lett., 2005, 5, 1658–1667 CrossRef CAS.
- G. Jones, V. Vullev, E. H. Braswell and D. Zhu, J. Am. Chem. Soc., 2000, 122, 388–389 CrossRef.
- B. R. Gibney, J. S. Johansson, F. Rabanal, J. J. Skalicky, A. J. Wand and P. L. Dutton, Biochemistry, 1997, 36, 2798–2806 CrossRef CAS.
- R. B. Hill, D. P. Raleigh, A. Lombardi and W. F. DeGrado, Acc. Chem. Res., 2000, 33, 745–754 CrossRef CAS.
- F. V. Cochran, S. P. Wu, W. Wang, V. Nanda, J. G. Saven, M. J. Therien and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 1346–1347 CrossRef CAS.
- J. Y. Shu, C. Tan, W. F. DeGrado and T. Xu, Biomacromolecules, 2008, 9, 2111–2117 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680 CAS.
- D. Y. Ryu, K. Shin, E. Drockenmuller, C. J. Hawker and T. P. Russell, Science, 2005, 308, 236–239 CrossRef CAS.
- Z. Q. Lin, D. H. Kim, X. D. Wu, L. Boosahda, D. Stone, L. LaRose and T. P. Russell, Adv. Mater., 2002, 14, 1373–1376 CrossRef CAS.
- J. Bang, B. J. Kim, G. E. Stein, T. P. Russell, X. Li, J. Wang, E. J. Kramer and C. J. Hawker, Macromolecules, 2007, 40, 7019–7025 CrossRef CAS.
- B. M. Discher, R. L. Koder, C. C. Moser and P. L. Dutton, Curr. Opin. Chem. Biol., 2003, 7, 741–748 CrossRef CAS.
- M. H. Hecht, a. Das, a. Go, L. H. Bradley and Y. N. Wei, Protein Sci., 2004, 13, 1711–1723 CrossRef CAS.
- D. E. Robertson, R. S. Farid, C. C. Moser, J. L. Urbauer, S. E. Mulholland, R. Pidikiti, J. D. Lear, A. J. Wand, W. F. Degrado and P. L. Dutton, Nature, 1994, 368, 425–431 CrossRef CAS.
- S. S. Huang, R. L. Koder, M. Lewis, A. J. Wand and P. L. Dutton, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5536–5541 CrossRef CAS.
- B. M. Discher, J. D. Lear, C. C. Moser and P. L. Dutton, Biophys. J., 2002, 82, 458A–458A.
- T. Xu, S. P. Wu, I. Miloradovic, M. J. Therien and J. K. Blasie, Nano Lett., 2006, 6, 2387–2394 CrossRef CAS.
- S. J. Singer, Adv. Protein Chem., 1963, 17, 1–68.
- J. T. Chin, S. L. Wheeler and A. M. Klibanov, Biotechnol. Bioeng., 1994, 44, 140–145 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- H. G. Borner and H. Schlaad, Soft Matter, 2007, 3, 394–408 RSC.
- O. D. Krishna and K. L. Kiick, Biopolymers, 2010, 94, 32–48 CrossRef CAS.
- J. T. Russell, Y. Lin, A. Boker, L. Su, P. Carl, H. Zettl, J. B. He, K. Sill, R. Tangirala, T. Emrick, K. Littrell, P. Thiyagarajan, D. Cookson, A. Fery, Q. Wang and T. P. Russell, Angew. Chem., Int. Ed., 2005, 44, 2420–2426 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- G. DeSantis and J. B. Jones, Curr. Opin. Biotechnol., 1999, 10, 324–330 CrossRef CAS.
- Y. Kodera, A. Matsushima, M. Hiroto, H. Nishimura, A. Ishii, T. Ueno and Y. Inada, Prog. Polym. Sci., 1998, 23, 1233–1271 CrossRef CAS.
- I. W. Hamley, A. Ansari, V. Castelletto, H. Nuhn, A. Rosler and H. A. Klok, Biomacromolecules, 2005, 6, 1310–1315 CrossRef CAS.
- B. R. Gibney, S. S. Huang, J. J. Skalicky, E. J. Fuentes, A. J. Wand and P. L. Dutton, Biochemistry, 2001, 40, 10550–10561 CrossRef CAS.
- Rf. Pasterna., G. C. Centuro, P. Boyd, L. D. Hinds, P. R. Huber, L. Francesc., P. Fasella, G. Engasser and E. Gibbs, J. Am. Chem. Soc., 1972, 94, 4511 CrossRef CAS.
- M. S. Choi, Tetrahedron Lett., 2008, 49, 7050–7053 CrossRef CAS.
- J. Strzalka, T. Xu, A. Tronin, S. P. Wu, I. Miloradovic, I. Kuzmenko, T. Gog, M. J. Therien and J. K. Blasie, Nano Lett., 2006, 6, 2395–2405 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |