DOI:
10.1039/C1SC00562F
(Edge Article)
Chem. Sci., 2011,
2, 2353-2360
Lectin-based electrochemical biosensor constructed by functionalized carbon nanotubes for the competitive assay of glycan expression on living cancer cells†
Received
15th August 2011
, Accepted 9th September 2011
First published on 22nd September 2011
Abstract
In this paper, we report a novel lectin-based electrochemical biosensor constructed by functionalized multiwalled carbon nanotubes (CNTs) for a competitive assay of glycan expression on living cells. The biosensor was fabricated by adsorbing poly(diallyldimethylammonium chloride) (PDDA)-functionalized CNTs (PDCNTs) onto a glassy carbon working electrode (GCE), followed by adsorbing glutathione (GSH)-protected gold nanoparticles (AuNPs). The resulting GCE/PDCNT/Au–GSH offers an effective platform for concanavalin A (Con A, a lectin) immobilization with high stability and bioactivity. A thiol-derivatized carbohydrate (thiomannosyl dimer) was synthesized to construct {CNT/thionine/Au–S–mannose} biocomposites, which exploit CNTs as carriers to load enormous amounts of thionine for generating an electrochemical signal and AuNPs for anchoring the thiomannosyl dimer. These two applications of the functionalized CNTs as a biosensing platform and biocomposites play important roles in signal enhancement for glycan detection. Using a competitive assay, the electrochemical biosensor shows good analytical performance with high sensitivity, selectivity and a rapid response for the analysis of mannose on human lung cancer cells as a model glycan. This proposed strategy possesses promising application for the assay of other glycans on living cancer cells with the selection of more lectins with the biosensor development.
Introduction
Glycans that are attached to glycoproteins on the cell surface play important roles in many biological events and their abnormal expression has been shown to correlate with some diseases, including diabetes, Alzheimer's disease, immune deficiencies and, in particular, cancers.1–3 The development of effective methods for the highly sensitive and accurate assay of glycans on living cells is critical to understand their roles in disease development and provides diagnostic tools to guide treatment. Although the reported approaches, such as mass spectrometry, nuclear magnetic resonance and chromatography, for glycan assay can reveal molecular details,4 they require expensive equipment, much time for sample preparation and are not amenable to living cell interrogation.5 Lectin microarrays have been developed as a glycan profiling tool to recognize cell surface glycans and most of the detection process involves the fluorescent labeling of cells.6,7 In recent years, electrochemical biosensors have been one of the excellent candidates for the analysis of biological components on living cells because of their simplicity, rapid response and potential ability for real-time and on-site detection.8,9 However, this remains a great challenge for the assay of glycans on living cells due to the lack of electroactive groups in glycans for signal generation. Most of the electrochemical biosensors for glycan analysis so far rely on enzymes as labels, which probably impedes their routine clinical applications due to high cost or relatively poor stability of enzymes.10 Therefore, many researchers are enthusiastic in exploiting new strategies for sensitive detection of cell surface glycans with limited amounts or low concentrations.11 At present, the competitive assay offers a promising solution in the evaluation of cell surface glycans for its good properties to promote the detection performance.
With the remarkable development of nanotechnology in materials science, the application of nanomaterials, such as nanotubes, nanoparticles and nanowires, has brought a great momentum for electrochemical biosensors and opens up new horizons for the highly sensitive detection of biological analytes.12–14 Among these nanomaterials, carbon nanotubes (CNTs) have attracted considerable interest due to their numerous signal amplification and excellent electrocatalytic effects.15–19 CNTs are fullerene-related structures that consist of graphite cylinders closed at either end with caps containing pentagonal rings.20 Owing to this structure, CNTs have many advantages in constructing electrochemical biosensors. For example, pioneering work has applied CNTs in electrode modification for the determination of a great many target analytes, such as dopamine,21 ascorbic acid,22 cytochrome c,23 catecholamine neurotransmitters24 and β-nicotinamide adenine dinucleotide (NADH).25 Meanwhile, CNTs have been used as efficient carriers to load a large amount of electroactive species (e.g., enzyme) as labels for amplifying the immunodetection of cancer biomarkers.26 Of late, research interest has been extended to functionalization of the surface of CNTs, e.g., with biomolecules and metal nanoparticles, through covalent or noncovalent methodologies.27–31 These functionalization strategies have generated a large variety of functional nanostructures with novel properties, which has been proven to be an effective way to enhance the solubility and biocompatibility of CNTs and to enrich the practical applications.
In this work, we fabricated the functionalized CNTs and demonstrated their application to the lectin-based electrochemical biosensor for the competitive assay of glycan expression on living cancer cells. Here, mannose present on human lung cancer cells (95-D and H1299) was used as a model glycan. Concanavalin A (Con A), a mannose-specific lectin with four binding sites for sugar epitopes, was used to prepare a Con A-based biosensor for mannose analysis. The biosensing platform was fabricated by first adsorbing poly(diallyldimethylammonium chloride) (PDDA)-functionalized CNTs (PDCNTs) onto a glassy carbon working electrode (GCE) and then adsorbing glutathione (GSH)-protected gold nanoparticles (AuNPs) by electrostatic adsorption for covalently linking Con A via amidization to the carboxylate groups of GSH on the AuNP surface (Scheme 2A). The GCE/PDCNT/Au–GSH not only offered an effective matrix for Con A attachment with high stability and bioactivity, but also played a critical role in signal enhancement in the following electrochemical detection. A thiol-derivatized mannose (thiomannosyl dimer: [mannose–S]2) was synthesized with two parts (Scheme 1):32,33 the disulfide portion [S–S] and the mannose portion, in which the disulfide part of thiomannosyl dimer anchored the molecule, while the mannose portion was responsible for competing with cell surface mannose to bind the biosensor surface-confined Con A.
 |
| | Scheme 1 The preparation of the thiomannosyl dimer and its assembly to AuNPs through a Au–S bond. | |
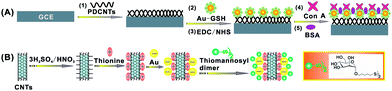 |
| | Scheme 2 A schematic representation of (A) the preparation procedure of the Con A-based biosensor and (B) the preparation of the {CNT/thionine/Au–S–mannose} biocomposites. | |
In view of signal enhancement, the {CNT/thionine/AuNP} was designed by the attachment of AuNPs to CNTs in the presence of thionine, which could effectively immobilize thiomannosyl dimer through a Au–S bond to form the {CNT/thionine/Au–S–mannose} biocomposites (Scheme 2B). After effective competition between the {CNT/thionine/Au–S–mannose} biocomposites and the cell surface mannose, the correlated response could be easily analyzed by detecting the immobilized thionine on the biosensor and a decreased peak current was obtained with an increasing cell concentration in the incubation solution. With the use of the functionalized CNTs as a biosensing platform and biocomposites for signal enhancement, the proposed strategy facilitates the highly sensitive detection of cell surface mannose and offers great promise for the analysis of other glycans on living cells by using a greater variety of lectins.
Results and discussion
Characterization of GSH-protected AuNPs
GSH-protected AuNPs were prepared for the purpose of covalently linking Con A via amidization to the carboxylate groups of GSH on the AuNP surface, which were characterized by transmission electron microscopy (TEM), UV-vis spectroscopy and Fourier transform infrared (FT-IR) spectra. As seen from TEM image, the spherical GSH-protected AuNPs were as dispersed particles with an average diameter of 3.5 nm (Fig. 1A). The spectra of the GSH-protected AuNPs exhibited a maximum absorption peak at ∼508 nm, while the AuNPs with a mean diameter of 18 nm showed the absorption band at ∼520nm (Fig. 1B). The presence of GSH on the AuNPs was also confirmed by the FT-IR spectra (Fig. 1C). As can be seen, GSH exhibited the characteristic peak at 2523 cm−1, assigned to the S–H stretching vibration. After conjugation of GSH on the AuNPs, the absorption band disappeared because of the formation of Au–S bond in GSH-protected AuNPs.
Characterization of the Con A-based biosensor
A scanning electron microscopy (SEM) technique was employed to confirm the fabrication process of the Con A-based biosensor. As shown in Fig. 2A, the PDCNTs homogeneously assembled on GCE were mostly in the form of small bundles and single tubes. Fig. 2B shows that numerous GSH-protected AuNPs were uniformly decorated onto the PDCNTs through the opposite-charged adsorption. This uniform nanostructure of the modified electrode could increase the effective surface area and also facilitate efficient Con A capture. When Con A was assembled on GCE/PDCNT/Au–GSH, the surface became much rougher and richer in texture (Fig. 2C). Owing to the porous structure and good biocompatibility, the PDCNT/Au–GSH provided an effective matrix for Con A binding and made the immobilized Con A hold high stability and bioactivity.34
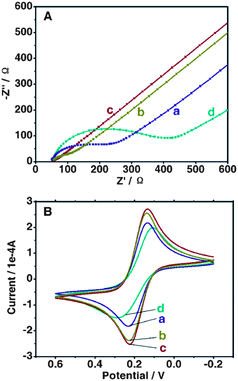
As a powerful tool for probing the interface features of surface-modified electrodes, electrochemical impedance spectroscopy (EIS) was used to study the electron-transfer resistance (Ret) changes of the electrode surface in the modification process. Fig. 3A illustrates the Nyquist plots of EIS for different modified electrodes in the presence of Fe(CN)63−/4− in PBS (pH 7.4 containing 0.10 M KCl). On bare GCE, the redox process of the probe showed a Ret of about 175 Ω (curve a), while the PDCNT-modified electrode exhibited a lower Ret for the redox probe (curve b). Subsequently, GSH-protected AuNPs were assembled on GCE/PDCNT and the resulting electrode exhibited an almost straight line (curve c), implying that GCE/PDCNT/Au–GSH could serve as an excellent transducer in promoting electron transfer. However, the attachment of Con A gave rise to a barrier towards the access of the redox probe to the electrode, resulting in an obvious increase in the Ret (GCE/PDCNT/Au–GSH/Con A, curve d). Cyclic voltammetry (CV) was also used to investigate the changes in the electrode behavior after each assembly step and the result was obviously consistent with that from EIS (Fig. 3B).
Preparation and characterization of CNT/thionine/Au
CNT/thionine/Au was fabricated by utilizing thionine as interlinkers and electroactive species, in which thionine enabled the negatively charged AuNPs to bind to the anionic CNT surface. Fig. 4A displays the TEM image of CNT/thionine/Au, which showed that AuNPs exhibited small clusters on CNTs. The observed clusters were well-separated and could be individually distinguished (the inset). UV-vis spectroscopy was used to investigate the preparation process of CNT/thionine/Au (Fig. 4B). As shown in curve b, CNT/thionine exhibited two absorption peaks at 610 nm and 280 nm. In comparison with those of pure thionine at 600 nm and 250 nm (curve a), the slight deviation between the absorption wavenumbers indicated an interaction between the CNTs and thionine. After the addition of AuNPs to CNT/thionine (curve c), the obvious red shift of the peak at 610 nm and blue shift of the peak at 280 nm provided the evidence for the successful formation of CNT/thionine/Au. Energy dispersive X-ray (EDX) analysis was also used to characterize the CNT/thionine/Au. The EDX spectra are presented in Fig. 4C, which reveals N and S peaks originating from adsorbed thionine and Au peaks attributable to the AuNPs.
Mechanism of the Con A-based electrochemical biosensor for the competitive assay of mannose expression on living cells
Scheme 3 illustrates the detailed mechanism of the electrochemical biosensor for the competitive assay of mannose expression on the cell surface. The Con A-based biosensor was exposed to an incubation solution containing the {CNT/thionine/Au–S–mannose} biocomposites and cancer cells, in which the biocomposites could compete effectively with cell surface mannose to recognize the biosensor surface-confined Con A. Subsequently, differential pulse voltammetry (DPV) was performed to detect thionine in the immobilized biocomposites on the biosensor. Owing to the competition of cell surface mannose with the {CNT/thionine/Au–S–mannose} biocomposites at a certain concentration, the amount of the immobilized biocomposites on the biosensor decreased with an increasing cell concentration in the recognition solution and, thus, a decreased current response was obtained. The current decrease was related to the amount of thionine, and thus reflected the expression level of cell surface mannose and the concentration of cancer cells in the incubation solution.
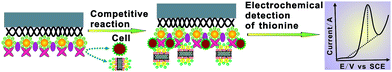 |
| | Scheme 3 A schematic representation of the mechanism of the electrochemical biosensor for the competitive assay of mannose expression on living cells. | |
Specific recognition of the {CNT/thionine/Au–S–mannose} biocomposites to the biosensor surface-confined Con A
The {CNT/thionine/Au–S–mannose} biocomposites were prepared to compete with cell surface mannose for binding to Con A. In order to demonstrate the specific recognition of the biocomposites to the biosensor surface-confined Con A, a blocking method was designed by first incubating the biosensor with the {CNT/thionine/Au–S–mannose} biocomposites for an electrochemical signal (Fig. 5). As shown in curve a, a high peak current was obtained, which is indicative of the successful binding of the biocomposites to the biosensor. Under the same experimental conditions, the Con A-based biosensor was initially incubated with a mannose sample and then the {CNT/thionine/Au-S-mannose} biocomposites. The peak current could hardly be detected (curve b), which was attributed to the fact that the initial incubation of the biosensor surface-confined Con A with mannose led to occupation of the Con A sites and prevented the subsequent binding of the {CNT/thionine/Au–S–mannose} biocomposites. The results indicated that the proposed strategy achieved the efficient recognition of the biocomposites toward the biosensor surface-confined Con A.
 |
| | Fig. 5 The DPV response on the Con A-based biosensor after incubation with (a) 0.025 μM of the {CNT/thionine/Au-S-mannose} biocomposites for 55 min and (b) 0.025 μM of a mannose sample for 55 min and then 0.025 μM of the biocomposites for 55 min. | |
Effects of the biocomposite concentration and incubation time on current response
In the competitive assay, the {CNT/thionine/Au–S–mannose} biocomposites were responsible for signal generation and greatly affected the current response. Fig. 6A shows the effect of the biocomposite concentration on the current response by saturation analysis. With an increasing biocomposite concentration, the current response increased and tended to a steady value after 0.025 μM, indicating the saturated binding of the biocomposites to the biosensor surface-confined Con A. Therefore, 0.025 μM was chosen as the optimal biocomposite concentration for the detection of cell surface mannose in the following experiments.
 |
| | Fig. 6 (A) The effect of the {CNT/thionine/Au–S–mannose} biocomposite concentration on the current response of the biosensor by saturation analysis. (B) The effect of the incubation time on the current response of the biosensor in the absence of cancer cells (a), in the presence of 95-D (b) and H1299 (c) at a cell concentration of 1.0 × 106 cells mL−1. | |
The time for the specific recognition between Con A and mannose was an important factor relevant to the sensitivity of the developed biosensor. Thus, the effect of the incubation time on the current response was investigated in the absence of cancer cells (a), in the presence of 95-D (b) and H1299 (c) at a cell concentration of 1.0 × 106 cells mL−1. As seen in Fig. 6B, the current response increased with the incubation time and started to level off after 55 min. Therefore, 55 min was selected as the incubation time for the specific recognition between Con A and mannose.
It is well-accepted that highly sensitive detection of living cells plays an important role in understanding disease development and in providing diagnostic tools to guide treatment.35 Hence, the Con A-based electrochemical biosensor was applied to detect cancer cells (95-D and H1299) based on the specific recognition between cell surface mannose and Con A and the obtained results were displayed in Fig. 7. As can be shown, the current response decreased with increasing 95-D (Fig. 7A) or H1299 (Fig. 7C) cell concentration in the recognition solution, which was attributed to the competition of cell surface mannose with the {CNT/thionine/Au–S–mannose} biocomposites to bind the biosensor surface-confined Con A. The calibration curve presented a linear relationship between the current response and the logarithm of cell concentrations ranging from 1.7 × 103 to 1.5 × 108 cells mL−1 for 95-D with a detection limit of 580 cells mL−1 (Fig. 7B) and from 25 to 1.0 × 106 cells mL−1 for H1299 with the detection limit of 12 cells mL−1 (Fig. 7D).
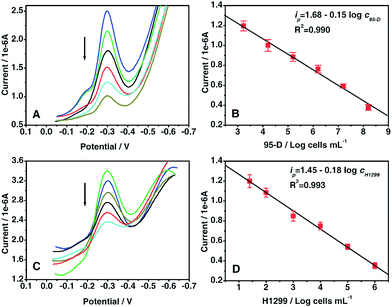 |
| | Fig. 7 DPV responses (A, C) and calibration curves (B, D) vs. the logarithm of the concentrations of 95-D cells (A, B) and H1299 cells (C, D). | |
When the GCE/PDCNT/Au–GSH/Con A-modified biosensor was stored in the refrigerator at 4 °C, the current response still remained at 90.1% of the initial response after a storage period of two weeks, showing a quite satisfying stability. The long-term stability might be attributed to the strong interactions between the PDCNT/Au–GSH and Con A. The reproducibility of the biosensor was also evaluated from the current response of the PDCNT/Au–GSH/Con A-modified electrode. A series of six measurements from the batch resulted in a relative standard deviation (RSD) of 5.2%, indicating that the reproducibility of the proposed biosensor was acceptable.
Moreover, the performance of the developed biosensor was compared with sensors reported in the literature. Table 1 summarizes the characteristics, such as the linear ranges and detection limits, for all of them. As can be seen, the proposed biosensor exhibited an attractive performance for the quantification of cancer cells with wide linear ranges and low detection limits.
Table 1 A comparison of the analytical performances of some biosensors for cell assay
Evaluation of mannose expression on cancer cells
In living systems, the recognition of cell surface mannose can provide useful tools for understanding its function in cancer development.40 Thus, the proposed biosensor was used to evaluate the amount of mannose on a single cell surface. To this purpose, a method was designed by first detecting the mannose sample to obtain the standard curve. As shown in Fig. 8A, the DPV response (ip) is proportional to the mannose concentration (cmannose) in the range 0.005–0.025 μM (R2 = 0.995). The linear regression equation was| | | ip (μA) = 1.11 − 6.45 cmannose (μM) | (1) |
 |
| | Fig. 8 Linear calibration plots of DPV peak current vs. concentrations of (A) mannose sample, (B) 95-D and (C) H1299 cells in Tris–HCl buffer. | |
Then, the electrochemical biosensor was used for the detection of cancer cells (95-D and H1299) at different concentrations. The current response (ip′) versus 95-D cell concentration (c95-D) showed a linear relation in the range 8.0 × 103–4.8 × 104 cells mL−1 (R2 = 0.991) (Fig. 8B). The regression equation was
| | | ip′ (μA) = 1.10 − 2.71 × 10−6c95-D | (2) |
Meanwhile, the ip′ for 95-D cell detection can be converted into the amount of mannose according to eqn (1), which represents mannose expression on 95-D cell surface. Consequently, with the use of eqn (1) and eqn (2), the amount of mannose on each cell surface (nmannose) can be calculated from the following equation:
where
ccell is the concentration of cancer
cells in
cells mL
−1. Four parallel measurements gave the average amount of
mannose on a single
cell surface to be 2.8 × 10
8 molecules for 95-D
cells. Using the same method, the amount of
mannose on each H1299
cell was also evaluated to correspond to 3.1 × 10
10 molecules (
Fig. 8C).
The mannose expression on living cells was evaluated based on the highly specific binding affinities between mannose and Con A. In order to investigate the selectivity of the Con A-based biosensor, it was used to detect 95-D cells (at a concentration of 2.0 × 104 cells mL−1, containing 0.0093 μM mannose) and a mannose sample (0.0093 μM) under the same experimental condition (Fig. S1, ESI†). The results indicated that the other biological molecules, such as amino acids and proteins on living cells, have no interference with the assay of cell surface mannose.
In addition, fluorescent analysis was also used to detect mannose expression on 95-D and H1299 cells by staining with Fluorescein Con A for the bioimaging experiment (Fig. S2, ESI†). Briefly, cancer cells were seeded on coverslips in 24-well plates (density 5000 cells/well) and cultured for 24 h. Afterwards, the cells were fixed using 4% paraformaldehyde for 5 min and washed with 0.10% Triton-X 100 in PBS three times. The fixed cells were incubated with Fluorescein Con A for 30 min in the dark at room temperature (RT). After being washed with PBS, the stained cells were observed under a fluorescence microscope at 10× and 20× magnifications. The results clearly demonstrated that mannose was more abundant in H1299 cells compared to 95-D cells, which supported the proposed methodology.
Compared with the existing methods for the evaluation of glycan expression on cell surfaces, this strategy offers the following advantages. (1) The proposed method is convenient and fast, particularly with much less demand on complicated equipment. (2) The procedure obviates the need for cell pretreatment, such as cell lysis and cell labeling, thus ensuring that the cells can be studied in their natural state. (3) The excellent electrical conductivity coupled with signal enhancement has the capability to enhance the sensitivity and can thus improve the detection limits and reduce the risk of false negative determination at low cell concentrations.
Conclusions
The present work describes the application of functionalized CNTs to design a Con A-based electrochemical biosensor for the competitive assay of mannose expression on living cancer cells. The biosensor applied GCE/PDCNT/AuNP–GSH to load Con A for the specific recognition of cell surface mannose. The {CNT/thionine/AuNP} was constructed to effectively immobilize a thiomannosyl dimer to form {CNT/thionine/Au–S–mannose} biocomposites for signal generation. With the use of functionalized CNTs as a biosensing platform and biocomposites for signal enhancement, the proposed strategy can analyze mannose expression with high sensitivity, selectivity and a rapid response. This facile biosensor has the potential to become a powerful tool for the study of other glycans on living cells by using a greater variety of lectins.
Experimental section
Materials and reagents
Reduced glutathione (GSH), poly(diallyldimethylammonium chloride) (PDDA), concanavalin A (Con A), bovine serum albumin (BSA), N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fluorescein Con A was obtained from Vector Laboratories (Burlingame, CA, USA). Multiwalled carbon nanotubes (CNTs, 10 nm–30 nm in diameter) were purchased from Nanotech Port Co., Ltd. (Shenzhen, China). Mannose was from Aladdin Reagent Database Inc. (Shanghai, China). HAuCl4, thionine and other chemical reagents were purchased from Chemical Reagents Co., Ltd. (Shanghai, China). All solutions were prepared with MilliQ water (18 MΩ·cm) from a Millipore system.
Human lung cancer cells: H1299 and 95-D cells were obtained from the Institute of Biomedical Sciences, East China Normal University (Shanghai, China). H1299 cells were maintained in DMEM medium with 10% fetal bovine serum at 37 °C in a humidified atmosphere of 5.0% CO2; 95-D cells were maintained in RPMI-1640 medium. At the logarithmic growth phase, cells were trypsinized and washed twice with phosphate buffer saline (PBS, pH 7.4) by centrifugation at 1000g for 5 min. The sediment was dispersed in Tris–HCl buffer (pH 7.4, containing 1.0 mM Mn2+, 1.0 mM Ca2+ and 0.10 M Na+) to obtain a homogeneous cell suspension.
Apparatus
Transmission electron microscopy (TEM) was collected on JEM-2100 (JEOL Ltd., Tokyo, Japan). UV-vis absorption spectra were recorded on a Cary 500 UV-vis–NIR spectrometer (Varian Inc., Palo Alto, CA, USA). Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet Nexus 670 instrument (Thermo Nicolet Co., USA) in KBr pellet form. Scanning electron microscopy (SEM) was carried out on S-4800 (Hitachi Co., Ltd., Tokyo, Japan) equipped with an energy dispersive X-ray (EDX) analyzer. Fluorescence images were collected by a fluorescence microscope (Leica DM4000B, Leica Microsystems, Nussloch, Germany). Electrochemical measurements were performed with a CHI 660C workstation (CH Instruments Inc., Austin, TX, USA) with a conventional three-electrode system comprised of a platinum wire as the auxiliary, a saturated calomel electrode (SCE) as the reference and a glassy carbon working electrode (GCE, 3 mm in diameter). Cyclic voltammetry (CV) was carried out at a scan rate of 100 mV s−1, and differential pulse voltammetry (DPV) at a pulse amplitude of 50 mV and a pulse width of 50 ms. Electrochemical impedance spectroscopy (EIS) was performed on an Autolab electrochemical analyzer (Eco Chemie, The Netherlands) using an alternating current voltage of 5.0 mV, within the frequency range 0.01 Hz–100 kHz.
Preparation of GSH-protected AuNPs
GSH-protected AuNPs were fabricated using a two-step procedure, which involves the adjustment of the pH of the GSH and HAuCl4 mixture with NaOH solution and the formation of AuNPs by the addition of NaBH4.41 Briefly, an aqueous solution of 1.0 mL HAuCl4 (0.024 M) was mixed with 7.8 mL of 0.019 M GSH solution. The pH of the resulting mixture was adjusted to ∼8.0 using 1.0 M NaOH. Then, a freshly prepared 2.0 mg mL−1 NaBH4 solution was added dropwise into the above solution with rapid stirring. After incubation for 2 h at room temperature (RT), the particle solution was transferred into a filter tube (50 KDa MW cutoff, Millipore) and purified by centrifuging at 3,500g and washing with water four times. Subsequently, the prepared GSH-protected AuNPs were redispersed in 3.0 mL water and stored in refrigerator at 4 °C.
Preparation of PDDA–CNTs (PDCNTs)
CNTs were carboxyl-functionalized and shortened by sonicating in a mixture of concentrated H2SO4/HNO3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) for 5 h, followed by extensive washing in water until the filtrate was neutral. The purified CNTs were functionalized with PDDA according to the following procedures: 0.50 mg mL−1 CNTs were dispersed into a 0.25% PDDA aqueous solution containing 0.50 M NaCl and the resulting dispersion was sonicated for 30 min to give a homogeneous black suspension.42,43 Residual PDDA was removed by high-speed centrifugation and the complex was rinsed three times with water to obtained PDDA-functionalized CNTs (PDCNTs). Then, the PDCNTs were redispersed in water with sonication to produce a solution, which was sonicated for 5 min immediately before preparing the biosensor.
:1 v/v) for 5 h, followed by extensive washing in water until the filtrate was neutral. The purified CNTs were functionalized with PDDA according to the following procedures: 0.50 mg mL−1 CNTs were dispersed into a 0.25% PDDA aqueous solution containing 0.50 M NaCl and the resulting dispersion was sonicated for 30 min to give a homogeneous black suspension.42,43 Residual PDDA was removed by high-speed centrifugation and the complex was rinsed three times with water to obtained PDDA-functionalized CNTs (PDCNTs). Then, the PDCNTs were redispersed in water with sonication to produce a solution, which was sonicated for 5 min immediately before preparing the biosensor.
Preparation of the Con A-based biosensor
GCE was used to fabricate the Con A-based biosensor. Prior to surface modification, the electrode was polished with 0.05 μm alumina followed by successive sonication in acetone, HNO3 (1:1 v/v), NaOH (50% w/w) and water. Afterwards, 10 μL of 5.0 mg mL−1 PDCNTs solution was dropped on the pretreated GCE and dried in a silica gel desiccator. The prepared GCE/PDCNT was immersed in GSH-protected AuNPs solution and incubated for 30 min to introduce the negatively charged Au-GSH. After being washed with water and dried with nitrogen, the GCE/PDCNT/Au–GSH was activated in a solution that contained 400 mM EDC and 100 mM NHS for 30 min and then incubated with 10 μL of 0.33 mg mL−1 Con A solution in 0.05% Tween-20/Tris–HCl buffer for 50 min. The obtained GCE/PDCNT/Au–GSH/Con A was further treated with 1.0% BSA/PBS for 30 min to block the nonspecific binding.
Synthesis of thiomannosyl dimer
The thiomannosyl dimer was prepared from mannose (M1) by the procedure shown in Scheme 1.32,33 M1 was treated with pyridine and acetic anhydride to give pentaacetyl mannose (M2). Pent-4-enyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside (M3) was synthesized by the glycosylation of M2 with 4-pentenyl alcohol in the presence of BF3–Et2O. M3 was treated with thioacetic acid and AIBN to obtain 5-thioacetoxypentyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl dimer (M4). Thiomannosyl dimer (M5) was obtained by deacylation of the acetyl form (M4) with MeONa. 1H NMR (500 MHz, CD3OD): δ 4.76 (d, J = 2 Hz, 1H), 3.84 (dd, J = 12, 2 Hz, 1H), 3.81 (dd, J = 3, 2 Hz, 1H), 3.70–3.78 (m, 3H), 3.63 (t, J = 10 Hz, 1H), 3.52–3.57 (m, 1H), 3.43–3.47 (m, 1H), 2.72 (t, J = 7.3 Hz, 2H), 1.71–1.78 (m, 2H), 1.61–1.67 (m, 2H), 1.49–1.56 (m, 2H); Maldi-TOF: [M + Na+]: 585.2.
Preparation of the {CNT/thionine/Au–S–mannose} biocomposites
The preparation procedures for the {CNT/thionine/Au–S–mannose} biocomposites are shown in Scheme 2B. An aqueous solution of 1.0 mL of the pre-treated CNTs (1.5 mg mL−1) was mixed with 1.0 mL of 3.0 mg mL−1 thionine solution and sonicated at RT for 12 h. The purple color of thionine disappeared and a black solution was obtained, suggesting that thionine was dissolved into the CNTs to form CNT/thionine through a π–π stacking force between two kinds of conjugated frames.44 Subsequently, 0.20 mL CNT/thionine solution was added dropwise into gold colloid (20 mL, which was prepared by the reduction of HAuCl4 with trisodium citrate45) under vigorous stirring. Immediately, the pink color faded and changed slowly into gray. After stirring for an addition 3 h, the resulting CNT/thionine/Au was isolated by centrifugation at 10,000g for 5 min and washed twice with water and then incubated with 0.50 mL of 10 mg mL−1 thiomannosyl dimer overnight under shaking to prepare the {CNT/thionine/Au–S–mannose} biocomposites. The biocomposites were isolated by centrifugation at 5000g for 10 min and washed twice with water. Then, Tris–HCl buffer (5.0 mL) was added to the prepared biocomposites to form a homogeneous dispersion and stored in a refrigerator at 4 °C.
Analytical procedure
The Con A-based biosensor was incubated with a mixture of 0.025 μM of the {CNT/thionine/Au–S–mannose} biocomposites and cancer cells at a given concentration for 55 min at RT (Tris–HCl buffer pH 7.4, containing 1.0 mM Mn2+, 1.0 mM Ca2+ and 0.10 M Na+). After the competitive binding between the {CNT/thionine/Au–S–mannose} biocomposites and cell surface mannose to the biosensor surface-confined Con A, the resulting electrode was immersed in HAc–NaAc buffer (pH 7.0) to detect the immobilized thionine on the biosensor with DPV from +0.10 V to −0.70 V (vs SCE).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (20775026, 21075041) and Science and Technology Commission of Shanghai Municipality (1052nm06500).
Notes and references
- A. Varki, Nature, 2007, 446, 1023–1029 CrossRef CAS.
- H. Stöckmann, A. A. Neves, S. Stairs, H. Ireland-Zecchini, K. M. Brindle and F. J. Leeper, Chem. Sci., 2011, 2, 932–936 RSC.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS.
- C. M. Szymanski, F. St. Michael, H. C. Jarrell, J. Li, M. Gilbert, S. Larocque, E. Vinogradov and J. R. Brisson, J. Biol. Chem., 2003, 278, 24509–24520 CrossRef CAS.
- K. T. Pilobello and L. K. Mahal, Curr. Opin. Chem. Biol., 2007, 11, 300–305 CrossRef CAS.
- A. Kuno, N. Uchiyama, S. Koseki-Kuno, Y. Ebe, S. Takashima, M. Yamada and J. Hirabayashi, Nat. Methods, 2005, 2, 851–856 CrossRef CAS.
- K. T. Pilobello, D. E. Slawek and L. K. Mahal, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11534–11539 CrossRef CAS.
- X. Zhang, Y. Teng, Y. Fu, L. Xu, S. Zhang, B. He, C. Wang and W. Zhang, Anal. Chem., 2010, 82, 9455–9460 CrossRef CAS.
- Y. L. Zhang, Y. Huang, J. H. Jiang, G. L. Shen and R. Q. Yu, J. Am. Chem. Soc., 2007, 129, 15448–15449 CrossRef CAS.
- J. Zhang, S. Song, L. Zhang, L. Wang, H. Wu, D. Pan and C. Fan, J. Am. Chem. Soc., 2006, 128, 8575–8580 CrossRef CAS.
- L. Ding, W. Cheng, X. J. Wang, S. J. Ding and H. X. Ju, J. Am. Chem. Soc., 2008, 130, 7224–7225 CrossRef CAS.
- X. Zhang, P. Geng, H. Liu, Y. Teng, Y. Liu, Q. Wang, W. Zhang, L. Jin and L. Jiang, Biosens. Bioelectron., 2009, 24, 2155–2159 CrossRef CAS.
- S. Bi, H. Zhou and S. Zhang, Chem. Sci., 2010, 1, 681–687 RSC.
- D. Du, L. Wang, Y. Shao, J. Wang, M. H. Engelhard and Y. Lin, Anal. Chem., 2011, 83, 746–752 CrossRef CAS.
- J.-J. Zhang, T.-T. Zheng, F.-F. Cheng and J.-J. Zhu, Chem. Commun., 2011, 47, 1178–1180 RSC.
- N. Li, R. Yuan, Y. Chai, S. Chen, H. An and W. Li, J. Phys. Chem. C, 2007, 111, 8443–8450 CAS.
- Y. J. Zhang, Y. Wen, Y. Liu, D. Li and J. H. Li, Electrochem. Commun., 2004, 6, 1180–1184 CrossRef CAS.
- P. Yu, Q. Qian, Y. Lin and L. Mao, J. Phys. Chem. C, 2010, 114, 3575–3579 CAS.
- H. Zhou, Z. Zhang, P. Yu, L. Su, T. Ohsaka and L. Mao, Langmuir, 2010, 26, 6028–6032 CrossRef CAS.
- X. Zhong, H.-J. Bai, J.-J. Xu, H.-Y. Chen and Y.-H. Zhu, Adv. Funct. Mater., 2010, 20, 992–999 CrossRef CAS.
- K. B. Wu, J. J. Fei and S. S. Hu, Anal. Biochem., 2003, 318, 100–106 CrossRef CAS.
- Z. Wang, J. Liu, Q. Liang, Y. Wang and G. Luo, Analyst, 2002, 127, 653–658 RSC.
- J. Wang, M. Li, Z. Shi, N. Li and Z. Gu, Anal. Chem., 2002, 74, 1993–1997 CrossRef CAS.
- J. Wang, M. Li, Z. Shi, N. Li and Z. Gu, Electroanalysis, 2002, 14, 225–230 CrossRef CAS.
- M. Musameh, J. Wang, A. Merkoci and Y. H. Lin, Electrochem. Commun., 2002, 4, 743–746 CrossRef CAS.
- X. Yu, B. Munge, V. Patel, G. Jensen, A. Bhirde, J. D. Gong, S. N. Kim, J. Gillespie, J. S. Gutkind, F. Papadimitrakopoulos and J. F. Rusling, J. Am. Chem. Soc., 2006, 128, 11199–11205 CrossRef CAS.
- J. P. Lei and H. X. Ju, Wiley Interdisciplinary Review, 2010, 2, 496–509 CAS.
- Z. J. Wang, M. Y. Li, Y. J. Zhang, J. H. Yuan, Y. F. Shen, L. Niu and A. Ivaska, Carbon, 2007, 45, 2111–2115 CrossRef CAS.
- Y. Wei, L.-T. Kong, R. Yang, L. Wang, J.-H. Liu and X.-J. Huang, Chem. Commun., 2011, 47, 5340–5342 RSC.
- Z. H. Wen, S. Q. Ci and J. H. Li, J. Phys. Chem. C, 2009, 113, 13482–13487 CAS.
- Q. Zhang, L. Zhang and J. H. Li, J. Phys. Chem. C, 2007, 111, 8655–8660 CAS.
- C. C. Lin, Y. C. Yeh, C. Y. Yang, C. L. Chen, G. F. Chen, C. C. Chen and Y. C. Wu, J. Am. Chem. Soc., 2002, 124, 3508–3509 CrossRef CAS.
- Y. K. Lyu, K. R. Lim, B. Y. Lee, K. S. Kim and W. Y. Lee, Chem. Commun., 2008, 4771–4773 RSC.
- S. J. Bao, C. M. Li, J. F. Zang, X. Q. Cui, Y. Qiao and J. Guo, Adv. Funct. Mater., 2008, 18, 591–599 CrossRef CAS.
- L. Ding, Q. Ji, R. Qian, W. Cheng and H. Ju, Anal. Chem., 2010, 82, 1292–1298 CrossRef CAS.
- H. L. Hu, H. Jiang, X. M. Wang and B. A. Chen, Electrochem. Commun., 2008, 10, 1121–1124 CrossRef CAS.
- M. Gu, J. Zhang, Y. Li, L. Jiang and J.-J. Zhu, Talanta, 2009, 80, 246–249 CrossRef CAS.
- J.-J. Zhang, F.-F. Cheng, T.-T. Zheng and J.-J. Zhu, Anal. Chem., 2010, 82, 3547–3555 CrossRef CAS.
- W. Cheng, L. Ding, J. P. Lei, S. J. Ding and H. X. Ju, Anal. Chem., 2008, 80, 3867–3872 CrossRef CAS.
- T. B. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. van Vliet, G. C. van Duijnhoven, J. Middel, I. L. Cornelissen, H. S. Nottet, V. N. Kewal-Ramani, D. R. Littman, C. G. Figdo and Y. van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS.
- V. Mani, B. V. Chikkaveeraiah, V. Patel, J. S. Gutkind and J. F. Rusling, ACS Nano, 2009, 3, 585–594 CrossRef CAS.
- R. J. Cui, H. P. Huang, Z. Z. Yin, D. Gao and J. J. Zhu, Biosens. Bioelectron., 2008, 23, 1666–1673 CrossRef CAS.
- R. J. Cui, C. Liu, J. M. Shen, D. Gao, J. J. Zhu and H. Y. Chen, Adv. Funct. Mater., 2008, 18, 2197–2204 CrossRef CAS.
- Z. J. Wang, M. Y. Li, P. P. Su, Y. J. Zhang, Y. F. Shen, D. X. Han, A. Ivaska and L. Niu, Electrochem. Commun., 2008, 10, 306–310 CrossRef CAS.
- K. C. Grabar, R. G. Freeman, M. B. Hommer and M. J. Natan, Anal. Chem., 1995, 67, 735–743 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.