Chemoselective intramolecular allylic C–H aminationversus C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C aziridination through Co(II)-based metalloradical catalysis†
C aziridination through Co(II)-based metalloradical catalysis†
Hongjian
Lu
,
Huiling
Jiang
,
Yang
Hu
,
Lukasz
Wojtas
and
X. Peter
Zhang
*
Department of Chemistry, University of South Florida, Tampa, FL 33620-5250, USA. E-mail: xpzhang@usf.edu; Fax: (+1) 813-974-3203; Tel: (+1) 813-974-7249
First published on 22nd September 2011
Abstract
Excellent chemoselectivity for intramolecular allylic C–H aminationversus C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aziridination was achieved through Co(II)-based metalloradical catalysis. Metalloradical catalyst [Co(P1)], the cobalt(II) complex of D2h-symmetric porphyrin 3,5-DitBu-IbuPhyrin, was shown to be highly effective for selective intramolecular allylic C–H amination of both N-bishomoallylic and N-allylic sulfamoyl azides. The Co(II)-catalyzed intramolecular 1,6-C–H amination of these azides provides a general and efficient method to access the synthetically useful allylic 1,3-diamines under neutral and non-oxidative conditions, without complication from the competitive olefin aziridination. The origin of this remarkable chemoselectivity has direct relevance to the radical mechanism of Co(II)-based metalloradical amination.
C aziridination was achieved through Co(II)-based metalloradical catalysis. Metalloradical catalyst [Co(P1)], the cobalt(II) complex of D2h-symmetric porphyrin 3,5-DitBu-IbuPhyrin, was shown to be highly effective for selective intramolecular allylic C–H amination of both N-bishomoallylic and N-allylic sulfamoyl azides. The Co(II)-catalyzed intramolecular 1,6-C–H amination of these azides provides a general and efficient method to access the synthetically useful allylic 1,3-diamines under neutral and non-oxidative conditions, without complication from the competitive olefin aziridination. The origin of this remarkable chemoselectivity has direct relevance to the radical mechanism of Co(II)-based metalloradical amination.
Introduction
Amination based on metal-catalyzed nitrene C–H insertion represents a powerful approach for the direct transformation of ubiquitous C–H bonds into valuable amine functionalities while offering potential control of various types of selectivities.1 When performed intramolecularly, the metal-catalyzed C–H amination may allow for a high control of regioselectivity by taking advantage of the preferred formation of certain ring sizes. Additionally, favorable entropy leads to an increase in reactivity. Efforts in this direction have been extremely fruitful and have led to the development of a number of successful catalytic systems for selective intramolecular C–H amination.1a,b Currently, the most recognized systems utilize dirhodium(II) tetracarboxylate-based catalysts in combination with different iminoiodane nitrene sources generated in situ from amide derivatives in the presence of terminal oxidants. The Rh(II)2-catalyzed intramolecular system has been shown to enable oxidative amination of both secondary and tertiary C–H bonds with excellent regioselectivity, as well as high stereoselectivity, producing synthetically useful 5- and 6-membered nitrogen heterocycles.1a,b Despite these achievements, the field faces several formidable challenges, among which is the control of chemoselectivity in intramolecular allylic C–H amination.1,2 Due to the electrophilic nature of the key metallonitrene intermediate involved in the Rh(II)2-catalyzed system, aziridination of more electron-rich CC π bonds typically favorably competes with amination of allylic C–H σ bonds.2–6 For example, Du Bois and coworkers recently reported that the intramolecular allylic C–H amination of N-Boc-N-bishomoallyl-sulfamide was out-competed by CC aziridination under the catalysis of [Rh2(esp)2] in combination with PhI(OAc)2 and MgO (eqn (1)).2j There have been a few reports on the selective intramolecular allylic C–H amination of sulfamates and carbamates.3 However, the yields of the desired allylic C–H amination products were typically low to moderate (43–60%).3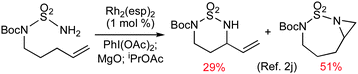 | (1) |
Departing from the widely-used Rh(II)2 and other closed-shell catalysts, we have been focusing our efforts on developing open-shell cobalt(II) porphyrin complexes ([Co(Por)]) as metalloradical catalysts for C–H amination.7,8 The [Co(Por)]-based metalloradical amination is unusual as it can effectively activate different azides as the nitrene sources, including sulfonyl,7acarbonyl,7bphosphoryl,7csulfamoyl7d and aryl8azides, under neutral and non-oxidative conditions.9,10 Consequently, the [Co(Por)]/azide catalytic system enjoys operational simplicity with a high functional group tolerance, as it obviates the need for terminal oxidants and other additives with nitrogen as the only byproduct. More importantly, the Co(II)-catalyzed reactions possess an uncommon capacity for efficient amination of strong primary C–H bonds. The unique profile of reactivity and selectivity is ascribed to the proposed radical mechanism of Co(II)-based metalloradical catalysis,7,11,12 which is in agreement with the result of a cyclopropane ring-opening experiment.7d On the basis of the anticipated radical mechanism, we envisioned a potential solution to address the issue of chemoselectivity in intramolecular allylic C–H amination, as illustrated with N-bishomoallylic sulfamoyl azides 1 (Scheme 1). In consideration of the relatively weak bond dissociation energy (∼83 kcal mol−1) of allylic C–H bonds together with the higher stability of allylic radical C compared to the alkyl radical B, the postulated key “nitrene radical” intermediate A is anticipated to proceed with H-atom abstraction rather than radical addition reaction although both processes would involve 7-membered transition states. Followed by an SHi (intramolecular homolytic substitution) reaction at the N-center, the C–H amination product 2 is expected to preferentially form in comparison to the CC aziridination product 3 (Scheme 1).
 | ||
| Scheme 1 A potential solution to achieve chemoselective intramolecular allylic C–H aminationversus CC aziridination based on proposed radical pathways of Co(II)-based metalloradical catalysis with azides. | ||
Results and discussion
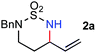
We began to test the metalloradical catalysis strategy for intramolecular allylic C–H amination with the model azide 1a (Scheme 2), which, along with other sulfamoyl azides, was readily prepared from the corresponding amine by a one-step procedure (see Supporting Information†).13,14 Under optimized conditions (2 mol % catalyst in C6H6 at 40 °C for 20 h), even common [Co(TPP)], which was shown to be ineffective for the amination of non-allylic/benzylic C–H bonds,7d could intramolecularly aminate one of the allylic C–H bonds of 1a to form the six-membered cyclic sulfamide 2a (Scheme 2), indicating a higher reactivity of allylic C–H bonds.15 Although the yield of 2a was moderate (43%), the catalytic reaction exhibited complete chemoselectivity for allylic C–H amination, as there was no observation of the competitive CC aziridination product 3a. When [Co(P1)], in which the D2h-symmetric porphyrin ligand 3,5-DitBu-IbuPhyrin P1 is attached with amide functionalities as hydrogen-bonding donors at the ortho-positions of the meso-phenyl groups,7c,d,16 was used as the catalyst, the reaction yield was dramatically improved to be near quantitative (99%) while retaining the complete chemoselectivity for allylic C–H amination (Scheme 2). It is worth noting the simplicity and cleanliness of the Co(II)/azide-based catalytic process. This high catalytic reactivity and excellent selectivity, together with the absence of oxidants and other additives, gave rise to a system where the desired allylic 1,3-diamine derivative 2a existed essentially as the sole compound after the reaction's completion (the only byproduct was nitrogen gas).

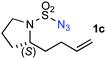
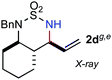
The [Co(P1)]-catalyzed intramolecular amination was shown to be applicable to sulfamoyl azides containing different types of allylic C–H bonds with varied substitution patterns (Table 1). As in the case of 1a, the secondary allylic C–H bonds in various azide derivatives, including 1b with homoallylic-methyl substitution, 1c with bishomoallylic substitution as part of the pyrrolidine ring and 1d–e with substitutions at both the homoallylic and bishomoallylic positions as part of the cyclohexane rings, could be chemoselectively aminated to produce the corresponding six-membered cyclic sulfamides 2a–e in excellent yields (entries 1–5). While the cyclic sulfamide 2b was formed anti-selectively (entry 2), it was noted that the formation of the 5,6-bicyclic structure in 2c was syn-selective, as confirmed by X-ray crystallographic analysis (entry 3; see Supporting Information†). Although the intramolecular amination reactions of both trans- and cis-cyclohexane-based azides 1d and 1e were found to be anti-selective, the diastereoselectivity (anti/syn > 95/5) for the formation of the 6,6-bicyclic structure 2e (entry 5) was shown to be higher than that (anti/syn 65/35) for the 6,6-bicyclic structure 2d (entry 4). The relative structures of both the isomers trans,cis-2d and cis,trans-2e were established by X-ray crystallographic analysis (see Supporting Information†).
| Entry | Sulfamoyl azide | Cyclic sulfamide | Yield (%)b |
|---|---|---|---|
| a Performed in C6H6 at 40 °C for 20 h using 2 mol % [Co(P1)] under N2 in the presence of 4 Å MS; [azide 1] = 0.10 M. b Isolated yields. c dr: 75/25. d dr: 25/75. e Confirmed by X-ray crystallographic analysis. f Purity of azide: ∼95%. g dr: 65/35. h dr: >95/5. i Contained <3% aziridination product. j Contained ∼5% (Z)-olefin compounds. k Contained ∼5% (E)-olefin product. | |||
| 1 |
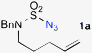
|
|
99% |
| 2 |

|

|
99% |
| 3 |
|

|
99% |
| 4 |

|
|
91% |
| 5 |

|
 ,
,
|
92% |
| 6 |
|
 ,
,
|
92% |
| 7 |

|
 ,
,
|
96% |
| 8 |

|

|
99% |
| 9 |

|

|
97% |
| 10 |

|

|
91% |
| 11 |

|

|
91% |
| 12 |

|

|
99% |
| 13 |

|
|
94% |
| 14 |
|

|
95% |
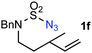
In addition to secondary allylic C–H bonds, the [Co(P1)]-based amination system was shown to be equally effective for tertiary allylic C–H bonds, as exemplified by the catalytic reaction of N-bishomoallylic sulfamoyl azide 1f (entry 6). Furthermore, even primary allylic C–H bonds could be successfully aminated in high reactivity and chemoselectivity. For example, the 1,1-disubstituted alkene-derived N-allylic sulfamoyl azide 1g was catalytically transformed to the desired six-membered cyclic sulfamide 2g in 96% yield viaamination of the methyl substituent (entry 7). Likewise, azides 1h and 1i derived from 1,2-disubstituted (E)-alkenes were also highly effective substrates for the Co(II)-catalyzed allylic 1,6-C–H amination (entries 8–9). When (Z)-alkene-containing azides 1j and 1k were employed as the substrates, the corresponding (Z)-cyclic sulfamides 2j and 2k were produced in 91% yields with concomitant formation of small amount of the (E)-isomers (entries 10–11), supporting the proposed radical mechanism (Scheme 1). As a representation of a substrate derived from a tri-substituted cyclic alkene, N-allylic sulfamoyl azide 1l was catalytically converted to the 6,6-bicyclic structure 2l in a near quantitative yield via a highly chemo- and regioselective allylic amination (entry 12).
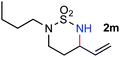
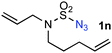
The outstanding chemoselectivity toward allylic C–H amination was further highlighted by the catalytic reactions of N-butyl-N-bishomoallyl-sulfamoyl azide 1m and N-allyl-N-bishomoallyl-sulfamoyl azide 1n (entries 13–14). It has been previously shown that the secondary non-allylic C–H bonds in N-butyl-sulfamoyl azides could also be effectively aminated by [Co(P1)] under similar conditions.7d Remarkably, the metalloradical catalyst could chemoselectively effect the intramolecular C–H amination of 1m only at the allylic position of its N-bishomoallyl group, without affecting the otherwise reactive secondary non-allylic C–H bonds of its N-butyl group, affording cyclic sulfamide 2m in 94% yield (entry 13). In the case of azide substrate 1n (entry 14), two different CC bonds exist that are both normally prone to intramolecular aziridination. Once again, it is extraordinary that [Co(P1)] could catalyze the chemoselective amination of the allylic C–H bonds without affecting the potentially reactive CC bonds. The corresponding cyclic sulfamide 2n bearing two alkene functionalities could be cleanly produced in 95% yield without observing any aziridination products (entry 14).
 | (2) |
 | (3) |
Due to its neutral and non-oxidative reaction conditions, the Co(II)-based metalloradical amination could tolerate substrates with various functional groups. For example, the intramolecular allylic C–H amination reactions of sulfamoyl azides 1o and 1p, which contain dimethylamino and cyano groups, respectively, could be properly catalyzed by [Co(P1)] to form the desired products 2o and 2p in high yields without affecting the functionalities (eqns (2) and (3)).
The reactivity and selectivity profiles of the Co(II)-catalyzed C–H amination accumulated from this and previous studies are in good agreement with the proposed radical mechanism of metalloradical catalysis (Scheme 1).7,11,12 To augment the supporting evidence from the prior cyclopropane ring-opening experiment,7d we designed azide substrate 1q, which was derived from a tri-substituted allylic amine, as a new type of radical probe for [Co(P1)]-catalyzed allylic C–H amination (Scheme 3). After azide 1q was subjected to the standard catalysis, the anticipated cyclic sulfamide 2q, which resulted from intramolecular amination of one of the primary allylic C–H bonds, was isolated in 90% yield. Its structure was confirmed by X-ray structural analysis (see Supporting Information†). While no aziridination product was observed, the cyclic sulfamide 2q′ was obtained as an additional product in 9% yield. It is apparent that product 2q′ is an isomer of product 2q generated from a rearrangement process. We take this result as more evidence to support the proposed radical mechanism of Co(II)-based metalloradical catalysis by invoking the key “Co(III)-nitrene radical” intermediate 1qA,11 which could be generated from azide 1q through metalloradical activation by [Co(P1)] after the release of N2 (Scheme 3). Further radical transfer from the Co-supported nitrogen radical 1qAvia1,6-hydrogen-atom abstraction of the allylic C–H bond would give rise to the corresponding allylic radical intermediate 1qB that can be represented by its two resonance forms 1qB′ and 1qB′′. As a result of the weak Co–N bond, the subsequent SHi (intramolecular homolytic substitution) reaction of the allylic radical is expected to proceed readily,11,17 leading to the formation of products 2q and 2q′ from resonance forms 1qB′ and 1qB′′, respectively. The predominant production of 2q over 2q′ is presumably a consequence of the relatively faster SHi rate of the primary allylic radical resonance form 1qB′ compared to that of the secondary allylic radical resonance form 1qB′′ (Scheme 3). Since the (Z)-isomer of 2q was not observed, it is concluded that isomerization of the allylic radical would have a high activation energy barrier.
 | (4) |
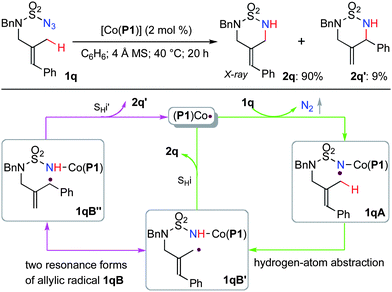 | ||
| Scheme 3 The utilization of allylic radical resonance to probe the radical mechanism of Co(II)-catalyzed intramolecular allylic C–H amination. | ||
Further evidence for the involvement of H-atom abstraction in the step-wise radical mechanism came from the study of the kinetic isotope effect (KIE). To this end, sulfamoyl azide 1r, in which one of two allylic hydrogen atoms is substituted with deuterium, was designed and synthesized for the experiment (eqn (4)). Under the catalytic conditions by [Co(P1)], both C–H and C–D amination products 2r-H and 2r-D were observed from the high-yielding reaction of the mono-deuterated substrate 1r. Analysis of the product mixture by 1H-NMR resulted in an intramolecular KIE of 6.2. This magnitude of primary KIE, which is much larger than the value of 1.9 measured for the Rh2-catalyzed intramolecular amination of benzylic C–H bonds,2k is in accord with the direct C–H bond breaking viaH-atom abstraction.
 | (5) |
The diverse array of cyclic sulfamides 2 bearing vinyl and other functionalities prepared by the Co(II)-catalyzed allylic C–H amination may find many applications in chemistry and biology.2,3,18 For example, cyclic sulfamide derivatives are widely investigated in medicinal chemistry as potential inhibitors for a number of important enzymes.19 Synthetically, they can function as useful precursors for the construction of valuable vinyl-substituted 1,3-diamines. To maintain the neutral and non-oxidative advantage of the amination process, we showed that the –SO2– bridging unit in the cyclic sulfamides 2 could be conveniently eliminated via transamination under neutral conditions through refluxing in 1,3-diaminopropane.20 For example, cyclic sulfamide 2a could be effectively converted to the corresponding diamine 4a in 95% yield in 3 h (eqn (5)).
Conclusions
In summary, we have demonstrated a novel approach based on the concept of metalloradical catalysis for addressing the challenging issue of chemoselectivity in intramolecular allylic C–H aminationversus competitive CC aziridination. The metalloradical catalyst [Co(P1)] has proved to be highly effective in activating both N-allylic and N-bishomoallylic sulfamoyl azides under neutral and non-oxidative conditions for the intramolecular amination of different types of allylic C–H bonds, including primary C–H bonds, with outstanding chemoselectivity. Evidence acquired from the studies of radical probe substrates suggests that the origin of the unusual reactivity and selectivity is likely attributed to the proposed radical mechanism,11,12 which involves the key “nitrene radical” intermediate processing with a preferred step-wise H-atom abstraction–homolytic substitution pathway. Efforts are under way to further expand the applications of Co(II)-based metalloradical catalysis for addressing other important issues in catalytic C–H amination.
Acknowledgements
We are grateful for financial support from the National Science Foundation (CHE-0711024).Notes and references
- (a) D. N. Zalatan and J. Du Bois, Top. Curr. Chem., 2010, 292, 347–378 CAS; (b) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061–5074 RSC; (c) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS; (d) H. M. L. Davies, Angew. Chem., Int. Ed., 2006, 45, 6422–6425 CrossRef CAS; (e) H. M. L. Davies and M. S. Long, Angew. Chem., Int. Ed., 2005, 44, 3518–3520 CrossRef CAS; (f) J. A. Halfen, Curr. Org. Chem., 2005, 9, 657–669 CrossRef CAS; (g) P. Muller and C. Fruit, Chem. Rev., 2003, 103, 2905–2919 CrossRef CAS.
- For select examples of intramolecular allylic C–H amination in competition with CC aziridination, see:
(a) F. Duran, L. Leman, A. Ghini, G. Burton, P. Dauban and R. H. Dodd, Org. Lett., 2002, 4, 2481–2483 CrossRef CAS;
(b) P. M. Wehn, J. H. Lee and J. Du Bois, Org. Lett., 2003, 5, 4823–4826 CrossRef CAS;
(c) K. W. Fiori, J. J. Fleming and J. Du Bois, Angew. Chem., Int. Ed., 2004, 43, 4349–4352 CrossRef CAS;
(d) C. Fruit and P. Muller, Tetrahedron: Asymmetry, 2004, 15, 1019–1026 CrossRef CAS;
(e) P. M. Wehn and J. Du Bois, Org. Lett., 2005, 7, 4685–4688 CrossRef CAS;
(f) C. J. Hayes, P. W. Beavis and L. A. Humphries, Chem. Commun., 2006, 4501–4502 RSC;
(g) M. Kim, J. V. Mulcahy, C. G. Espino and J. Du Bois, Org. Lett., 2006, 8, 1073–1076 CrossRef CAS;
(h) J. J. Fleming, M. D. McReynolds and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 9964–9975 CrossRef CAS;
(i) D. E. Olson and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 11248–11249 CrossRef CAS;
(j) T. Kurokawa, M. Kim and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 2777–2779 CrossRef CAS;
(k) K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042–3051 CrossRef CAS;
(l) L. A. Boralsky, D. Marston, R. D. Grigg, J. C. Hershberger and J. M. Schomaker, Org. Lett., 2011, 13, 1924–1927 CrossRef CAS.
- For examples of selective intramolecular allylic C–H amination, see: (a) D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 9220–9221 CrossRef CAS; (b) E. Milczek, N. Boudet and S. Blakey, Angew. Chem., Int. Ed., 2008, 47, 6825–6828 CrossRef CAS; (c) H. Lebel, K. Huard and S. Lectard, J. Am. Chem. Soc., 2005, 127, 14198–14199 CrossRef CAS.
- For intermolecular allylic C–H amination, see: (a) J. P. Mahy, G. Bedi, P. Battioni and D. Mansuy, Tetrahedron Lett., 1988, 29, 1927–1930 CrossRef CAS; (b) I. Nageli, C. Baud, G. Bernardinelli, Y. Jacquier, M. Moran and P. Muller, Helv. Chim. Acta, 1997, 80, 1087–1105 CrossRef CAS; (c) X. Q. Yu, J. S. Huang, X. G. Zhou and C. M. Che, Org. Lett., 2000, 2, 2233–2236 CrossRef CAS; (d) Y. Kohmura and T. Katsuki, Tetrahedron Lett., 2001, 42, 3339–3342 CrossRef CAS; (e) J. L. Liang, J. S. Huang, X. Q. Yu, N. Y. Zhu and C. M. Che, Chem.–Eur. J., 2002, 8, 1563–1572 CrossRef CAS; (f) C. G. Liang, F. Robert-Pedlard, C. Fruit, P. Muller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641–4644 CrossRef CAS; (g) C. G. Liang, F. Collet, F. Robert-Peillard, P. Muller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343–350 CrossRef CAS; (h) F. Collet, C. Lescot, C. G. Liang and P. Dauban, Dalton Trans., 2010, 39, 10401–10413 RSC.
- For representative examples of Pd-catalyzed allylic C–H aminationvia non-nitrene C–H activation, see: (a) G. S. Liu and Y. C. Wu, Top. Curr. Chem., 2010, 292, 195–209 CAS; (b) K. J. Fraunhoffer and M. C. White, J. Am. Chem. Soc., 2007, 129, 7274–7276 CrossRef CAS; (c) S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316–3318 CrossRef CAS; (d) G. S. Liu, G. Y. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733–4736 CrossRef CAS; (e) G. Y. Yin, Y. C. Wu and G. S. Liu, J. Am. Chem. Soc., 2010, 132, 11978–11987 CrossRef CAS; (f) H. F. Du, W. C. Yuan, B. G. Zhao and Y. Shi, J. Am. Chem. Soc., 2007, 129, 7496–7497 CrossRef CAS; (g) H. F. Du, B. G. Zhao and Y. Shi, J. Am. Chem. Soc., 2008, 130, 8590–8591 CrossRef CAS; (h) R. I. McDonald and S. S. Stahl, Angew. Chem., Int. Ed., 2010, 49, 5529–5532 CrossRef CAS.
- For select example of other types of metal-catalyzed allylic C–H amination, see: (a) R. S. Srivastava and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 3302–3310 CrossRef CAS; (b) R. S. Srivastava, N. R. Tarver and K. M. Nicholas, J. Am. Chem. Soc., 2007, 129, 15250–15258 CrossRef CAS; (c) M. Johannsen and K. A. Jorgensen, J. Org. Chem., 1994, 59, 214–216 CrossRef CAS.
- (a) J. V. Ruppel, R. M. Kamble and X. P. Zhang, Org. Lett., 2007, 9, 4889–4892 CrossRef CAS; (b) H. J. Lu, V. Subbarayan, J. R. Tao and X. P. Zhang, Organometallics, 2010, 29, 389–393 CrossRef CAS; (c) H. J. Lu, J. R. Tao, J. E. Jones, L. Wojtas and X. P. Zhang, Org. Lett., 2010, 12, 1248–1251 CrossRef CAS; (d) H. J. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192–10196 CrossRef CAS.
- For works by Cenini and coworkers on [Co(Por)]-catalyzed intermolecular C–H amination with aryl azides, see refs 10a–e and: (a) S. Cenini, E. Gallo, A. Penoni, F. Ragaini and S. Tollari, Chem. Commun., 2000, 2265–2266 RSC; (b) F. Ragaini, A. Penoni, E. Gallo, S. Tollari, C. L. Gotti, M. Lapadula, E. Mangioni and S. Cenini, Chem.–Eur. J., 2003, 9, 249–259 CrossRef.
- For reviews on the use of azides as nitrene sources for metal-catalyzed nitrene transfers, see: (a) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831–3846 RSC; (b) S. Cenini, E. Gallo, A. Caselli, F. Ragaini, S. Fantauzzi and C. Piangiolino, Coord. Chem. Rev., 2006, 250, 1234–1253 CrossRef CAS; (c) T. Katsuki, Chem. Lett., 2005, 34, 1304–1309 CrossRef CAS.
- For metal-catalyzed C–H amination with azides, see refs 7, 8 and: (a) S. Cenini, S. Tollari, A. Penoni and C. Cereda, J. Mol. Catal. A: Chem., 1999, 137, 135–146 CrossRef CAS; (b) A. Caselli, E. Gallo, F. Ragaini, F. Ricatto, G. Abbiati and S. Cenini, Inorg. Chim. Acta, 2006, 359, 2924–1932 CrossRef CAS; (c) A. Caselli, E. Gallo, S. Fantauzzi, S. Morlacchi, F. Ragaini and S. Cenini, Eur. J. Inorg. Chem., 2008, 3009–3019 CrossRef CAS; (d) D. Intrieri, A. Caselli, F. Ragaini, S. Cenini and E. Gallo, J. Porphyrins Phthalocyanines, 2010, 14, 732–740 Search PubMed; (e) S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, N. Casati, P. Macchi and S. Cenini, Chem. Commun., 2009, 3952–3954 RSC; (f) B. J. Stokes, H. J. Dong, B. E. Leslie, A. L. Pumphrey and T. G. Driver, J. Am. Chem. Soc., 2007, 129, 7500–7501 CrossRef CAS; (g) H. Dong, M. Shen, J. E. Redford, B. J. Stokes, A. L. Pumphrey and T. G. Driver, Org. Lett., 2007, 9, 5191–5194 CrossRef CAS; (h) M. H. Shen, B. E. Leslie and T. G. Driver, Angew. Chem., Int. Ed., 2008, 47, 5056–5059 CrossRef CAS; (i) M. Shen and T. G. Driver, Org. Lett., 2008, 10, 3367–3370 CrossRef CAS; (j) B. J. Stokes, B. Jovanovic, H. J. Dong, K. J. Richert, R. D. Riell and T. G. Driver, J. Org. Chem., 2009, 74, 3225–3228 CrossRef CAS; (k) B. J. Stokes, K. J. Richert and T. G. Driver, J. Org. Chem., 2009, 74, 6442–6451 CrossRef CAS; (l) K. Sun, R. Sachwani, K. J. Richert and T. G. Driver, Org. Lett., 2009, 11, 3598–3601 CrossRef CAS; (m) Y. G. Liu and C. M. Che, Chem.–Eur. J., 2010, 16, 10494–10501 CrossRef CAS; (n) Y. G. Liu, J. H. Wei and C. M. Che, Chem. Commun., 2010, 46, 6926–6928 RSC; (o) K. Omura, M. Murakami, T. Uchida, R. Irie and T. Katsuki, Chem. Lett., 2003, 32, 354–355 CrossRef CAS; (p) Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961–9964 CrossRef CAS; (q) W. G. Shou, J. A. Li, T. X. Guo, Z. Y. Lin and G. C. Jia, Organometallics, 2009, 28, 6847–6854 CrossRef CAS.
- For studies on the radical mechanism of [Co(Por)]-catalyzed C–H amination involving Co(III)-nitrene radical intermediates, see: (a) V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264–12273 Search PubMed. For related DFT studies on the radical mechanism of [Co(Por)]-catalyzed aziridination, see: (b) A. I. Olivos Suarez, H. Jiang, X. P. Zhang and B. de Bruin, Dalton Trans., 2011, 40, 5697–5705 RSC; (c) K. H. Hopmann and A. Ghosh, ACS Catal., 2011, 1, 597–600 Search PubMed.
- For studies on the radical mechanism of [Co(Por)]-catalyzed cyclopropanation involving Co(III)-carbene radical intermediates, see: (a) W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891–10902 CrossRef CAS; (b) J. L. Belof, C. R. Cioce, X. Xu, X. P. Zhang, B. Space and H. L. Woodcock, Organometallics, 2011, 30, 2739–2746 Search PubMed; (c) H. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 Search PubMed.
- (a) J. Griffith, J. Chem. Soc. C, 1971, 3191–3195 RSC; (b) W. L. Matier, W. T. Comer and D. Deitchman, J. Med. Chem., 1972, 15, 538–541 CrossRef CAS; (c) E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS.
- Sulfamoyl azides were reported to be chemically stable, even in strong acidic and basic conditions. (see ref. 13) Our DSC experiments indicated that these sulfamoyl azides were thermally stable without decomposition up to at least 100 °C; see Supporting Information for a representative DSC plot†.
- In the absence of a catalyst, there was no reaction. Simple Co(II) compounds, such as CoCl2 and Co(OAc)2, were found to have no catalytic activity even using 20 mol % catalyst loadings.
- J. V. Ruppel, J. E. Jones, C. A. Huff, R. M. Kamble, Y. Chen and X. P. Zhang, Org. Lett., 2008, 10, 1995–1998 CrossRef CAS.
- It was found that the addition of radical traps, such as 20 mol % hydroquinone or TEMPO, had no significant effect on the catalytic C–H amination of azide 1a. This result is consistent with the very low barrier (or barrierless) of the SHi step suggested by the DFT calculations (see ref. 11).
- For other select references on cyclic sulfamides, see: (a) K. C. Nicolaou, D. A. Longbottom, S. A. Snyder, A. Z. Nalbandian and X. Huang, Angew. Chem., Int. Ed., 2002, 41, 3866–3870 Search PubMed; (b) Z. Rgania, J.-Y. Winum, F.-Z. Smaine, L. Toupet, N.-E. Aouf and J.-L. Montero, Tetrahedron, 2003, 59, 6051–6056 Search PubMed; (c) P. D. Johnson, S. A. Jewell and D. L. Romero, Tetrahedron Lett., 2003, 44, 5483–5485 Search PubMed; (d) K. C. Nicolaou, S. A. Snyder, D. A. Longbottom, A. Z. Nalbandian and X. Huang, Chem.–Eur. J., 2004, 10, 5581–5606 CrossRef CAS; (e) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef CAS; (f) T. P. Zabawa, D. Kasi and S. R. Chemler, J. Am. Chem. Soc., 2005, 127, 11250–11251 CrossRef CAS; (g) T. P. Zabawa and S. R. Chemler, Org. Lett., 2007, 9, 2035–2038 CrossRef CAS; (h) K. Muniz, J. Streuff, C. H. Hvelmann and A. Nunez, Angew. Chem., Int. Ed., 2007, 46, 7125–7127 CrossRef CAS; (i) S. R. Chemler, J. Organomet. Chem., 2011, 696, 150–158 Search PubMed; (j) R. G. Cornwall, B. G. Zhao and Y. Shi, Org. Lett., 2011, 13, 434–437 Search PubMed.
- (a) A. B. Reitz, G. R. Smith and M. H. Parker, Expert Opin. Ther. Pat., 2009, 19, 1449–1453 Search PubMed; (b) J. Y. Winum, A. Scozzafava, J. L. Montero and C. T. Supuran, Expert Opin. Ther. Pat., 2006, 16, 27–47 Search PubMed.
- R. B. C. Jagt, P. Y. Toullec, D. Geerdink, J. G. de Vries, B. L. Feringa and A. D. J. Minnaard, Angew. Chem., Int. Ed., 2006, 45, 2789–2791 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for new compounds; CIF files of compounds 2c, 2d, 2e, 2q. CCDC reference numbers 830353–830356. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00366f |
| This journal is © The Royal Society of Chemistry 2011 |