Chemical synthesis and functionalization of clickable glycosylphosphatidylinositol anchors†
Benjamin M.
Swarts
and
Zhongwu
Guo
*
Department of Chemistry, Wayne State University, 5101 Cass Avenue, Detroit, Michigan 48202, USA. E-mail: zwguo@chem.wayne.edu; Fax: +1-313-577-8822; Tel: +1-313-577-2557
First published on 6th September 2011
Abstract
Glycosylphosphatidylinositol (GPI) anchorage is a common posttranslational modification of eukaryotic proteins. Chemical synthesis of structurally defined GPIs and GPI derivatives is a necessary step toward understanding the properties and functions of these molecules in biological systems. In this work, the synthesis of several functionalized GPI anchors was accomplished using the para-methoxybenzyl (PMB) group for permanent hydroxyl protection, which allowed the incorporation of functionalities that are incompatible with permanent protecting groups traditionally used in carbohydrate synthesis. A flexible convergent–divergent assembly strategy enabled efficient access to a diverse set of target structures, including “clickable” alkyne- and azide-modified GPIs. For global deprotection, a one-pot reaction was employed to afford the target GPIs in excellent yields (85–97%). Fully deprotected clickable GPI derivatives were readily conjugated to imaging and affinity probes via Cu(I)-catalyzed and Cu-free strain-promoted [3 + 2] cycloaddition to result in GPI-Flour and GPI-Biotin conjugates, respectively.
Introduction
The attachment of membrane proteins and glycoproteins to the cell surface by glycosylphosphatidylinositol (GPI) anchors, a structurally diverse class of glycolipids, is ubiquitous in eukaryotic species.1–4 To date, more than 250 eukaryotic membrane proteins have been established as GPI-anchored,5 and a recent genomic study has suggested that as many as 0.5–1% of all eukaryotic proteins may be anchored to the cell surface by GPIs.6 GPIs and GPI-anchored molecules play vital roles in numerous biological and pathological processes, such as cell recognition and adhesion,7 signal transduction,8,9 pathogenic infections,10 enzymatic reactions on the cell surface,11 and functioning as cellular markers.4The discovery of GPI anchorage as a unique mode of membrane-protein binding spanned the 1970s and 1980s1 and culminated in the full characterization of the Trypanosoma brucei variant surface glycoprotein GPI anchor in 1988 by Ferguson and co-workers.12 Since then, numerous GPIs have been identified and characterized, and a highly conserved core structure has been established: H2NEt-(P)-6-Manα(1 → 2)Manα(1 → 6)Manα(1 → 4)-GlcNH2α(1 → 6)myo-inositol-1-(P)-glycerolipid (Fig. 1). The protein C-terminus is covalently linked to the non-reducing end of the GPI core glycan through a phosphoethanolamine bridge, and the entire structure is anchored to the cell surface by insertion of the phosphatidylinositol (PI) fatty acid chains into the membrane bilayer. The tetrasaccharide core, whose function remains obscure, exhibits considerable structural diversity amongst known GPIs, primarily in the form of additional carbohydrate and phosphoethanolamine units linked to various positions. For example, mannose and galactose mono/oligomers are commonly appended to the Man-III 2-O-position and the Man-I 3/4-O-positions, respectively, while the Man-I 2-O-position frequently carries a phosphoethanolamine group.4 Additionally, the PI moiety may be palmitoylated at the inositol 2-O-position, while its phosphoglycerolipid can undergo fatty acid remodeling,13 leading to modified lipids that can be acyl- or alkyl-linked or exhibit varying chain length and unsaturation patterns.
 | ||
| Fig. 1 GPI structure and anchoring function. | ||
Despite impressive advances in elucidating the biosynthesis, structure, and function of GPI anchors and GPI-anchored proteins,14 many aspects of these molecules remain poorly understood. For example, the significance of structural complexity and diversity of GPI anchors, particularly in the conserved glycan core, is essentially unexplained and prompts structure–activity relationship studies. The use of GPI partial structures in the development of anti-malarial vaccines15 suggests GPIs have therapeutic value, but new technologies are needed to explore their potential more fully. Imaging of GPI anchors in vivo could provide insight into their expression, distribution, and endocytosis. Another emerging area of research is “GPIomics,” which seeks to develop proteomic tools for identifying proteins carrying the GPI anchor as a posttranslational modification.
To accelerate progress in these and other areas of GPI research, homogeneous and structurally defined GPIs and GPI derivatives must be accessible in sufficient purity and quantity, a requirement that can be addressed by chemical synthesis. To date, several research groups, including ours, have completed total syntheses of GPI anchors and related partial structures.15–35 However, very little work has been devoted to the synthesis of functionalized GPIs that are chemically modified to facilitate biological studies. Seeberger and co-workers reported the synthesis of a GPI derivative containing a cysteine residue to enable the chemoselective attachment of prion protein via native chemical ligation.34 The Bertozzi group synthesized a series of truncated unnatural GPI analogs carrying green fluorescent protein as an imaging motif and evaluated the behavior of the compounds in supported lipid bilayers and live cells.36,37 In this edge article, we describe the development of a synthetic strategy that enabled the preparation of a panel of functionalized GPI anchors containing unprecedented functional versatility. The target GPIs, which exhibit naturally occurring core and PI structures, were outfitted with alkyne and azide tags to facilitate Cu(I)-catalyzed and bioorthogonal click chemistry, as well as fluorescent and affinity tags to enable imaging and proteomics applications.
Results and discussion
Synthetic targets and strategy
A series of GPI anchors (1–5) modified with functionalities such as alkyne, azide, fluorescent, and biotin tags was designed as a broadly applicable toolset for the study of GPIs (Fig. 2). Our primary targets were clickable GPIs 1–3, which carried small, bioorthogonal alkyne or azide groups to allow control of GPI labeling with functional probes. Because imaging and affinity probes are often large, hydrophobic, and potentially perturbing to biological systems, the ability to chemoselectively attach them via click chemistry with temporal control is an important feature. For the site of functionalization, the Man-I 2-O-position was chosen for two primary reasons: (1) as discussed above, it is a common point of modification in naturally occurring GPIs, so we did not expect significant structural or functional consequences; (2) orthogonal protection of the Man-I 2-O-position was synthetically feasible and also required38 for the key glycosylation reaction of our synthetic strategy. We initially selected Alkynyl-GPI 1 bearing an acyl-linked alkyne group to serve as a click tag. After discovering that the acyl-based linker was prone to cleavage during storage, we designed second generation targets Alkynyl-GPI 2 and Azido-GPI 3, which possessed more stable alkyl-based linkers. To demonstrate that these GPIs could be further derivatized via click chemistry, fully deprotected GPIs 2 and 3 were then coupled to imaging and affinity probes to obtain GPI-Fluor 4 and GPI-Biotin 5, respectively.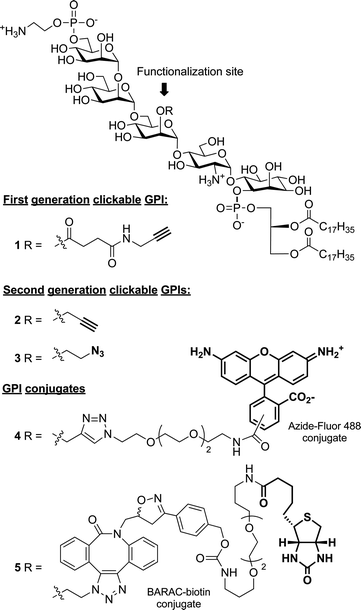 | ||
| Fig. 2 Synthetic targets. | ||
The target GPIs 1–5 presented a considerable synthetic challenge. Most GPI syntheses reported to date have employed the benzyl group for permanent hydroxyl protection due to ease of installation, stability, and mild deprotection. However, global deprotection of benzyl groups using Pd-catalyzed hydrogenolysis is incompatible with alkynes, azides, alkenes, thiols, thioethers, and other potentially reducible or catalyst-poisoning functional groups. This limitation prevents the incorporation of functionalities for click chemistry and many imaging and affinity moieties and was therefore quite apparent to us at the outset of this project. On the other hand, base-sensitive groups such as esters and peptides/glycopeptides in the target molecule are a liability when acyl-based permanent protection is used. To address this issue, our lab has recently explored the strategy of using the para-methoxybenzyl (PMB) protecting group, which can be removed under acidic or oxidative conditions, for permanent hydroxyl protection in the synthesis of functionalized GPI anchors. As a proof-of-principle, we first applied this strategy to the synthesis of a GPI anchor bearing unsaturated lipid chains, which was highlighted by a three-step, one-pot global deprotection that afforded the target compound in 81% yield.35 Here, we provide a full account of the development of the PMB protection strategy and its application to the synthesis of functionalized GPI anchors 1–5.
Retrosynthetic analysis (Scheme 1) suggested a convergent–divergent strategy that would optimize efficiency while enabling access to a diverse set of GPIs. Flexibility was emphasized in the synthetic design to accommodate the structural heterogeneity of naturally occurring GPIs, particularly in the PI lipid chains, as well as our desire to functionalize GPIs for biological study. Accordingly, pseudodisaccharide alcohol 6 and trimannose thioglycoside 7 were targeted as adaptable PMB-protected intermediates that would allow for choice of phosphoglycerolipid structure and functional tag in the target GPI. Pseudodisaccharide alcohol 6 would arise from an α-stereoselective glycosylation between optically pure inositol 8 and glucosaminyl donor 9. Trimannoside 7, which contained an orthogonal allyl group at the Man-I 2-O-position, the site of functionalization, would be built from mannose monomers 10–12 using sequential α-stereoselective glycosylations. In this edge article, we discuss the construction of common intermediates 6 and 7 followed by their elaboration to functionalized GPI anchors 1–5.
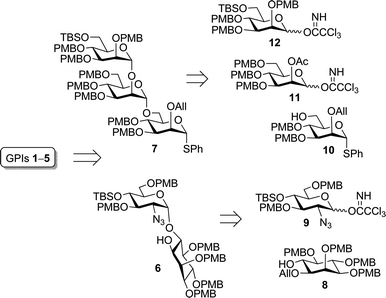 | ||
| Scheme 1 Convergent–divergent synthetic strategy. | ||
Synthesis of pseudodisaccharide 6
The construction of pseudodisaccharide 6 presented two primary challenges, including the preparation of an optically pure inositol derivative and the formation of the glucosamine-inositol α-glycosidic bond, which is notoriously difficult to form in high stereoselectivity in the context of GPI synthesis.39 To accomplish the former task, inositol derivative 8 was prepared using a delayed 1,6-O-differentiation route in combination with chemical enantiomeric resolution. To achieve α-glycosylation with acceptable selectivity in the pseudodisaccharide formation, 2-azido-2-deoxy-D-glucosyl donors were employed as latent glucosamine units due to the non-participating property of the 2-azido group.2,3:4,5-Di-O-cyclohexylidene-myo-inositol (13) was selected as a starting point for the preparation of 1,6-O-differentiated inositol 8. Our group has previously synthesized analogous benzyl-protected inositols40via initial 1,6-O-differentiation of 13 invoked by a stannylene acetal-directed selective allylation, which favored the 6-O-position in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to give compound 14 in high yield (Scheme 2A). Translation of this strategy to a suitable PMB-protected inositol derivative was halted because we were unable to identify a common orthogonal protecting group (PG) for subsequent protection of the 1-O-position that satisfied the following criteria: (1) PG must not be susceptible to cleavage or migration by strong acid (AcCl/MeOH) or base (NaH/PMBCl); (2) PG must be orthogonal to PMB, allyl, tert-butyldimethylsilyl (TBS), and azido groups. Therefore, a new route based on delayed 1,6-O-differentiation was devised (Scheme 2B). The 1-O- and 6-O-positions of racemic diol (±)-13 were simultaneously protected as allyl ethers, which were unaffected by the following cyclohexylidene removal and para-methoxybenzylation reactions. Pd-catalyzed deallylation of bis-allyl ether (±)-17 was inefficient (yield < 50%), so alternative methods were explored. Cha's protocol, which employs Ti(Oi-Pr)4 and a commercially available Grignard reagent,41 cleanly removed both allyl groups to provide inositol (±)-18 in 85% yield. For the delayed 1,6-O-differentiation of PMB-protected diol (±)-18, we expected that the regioselectivity of stannylene acetal-directed alkylation would fortuitously invert with respect to cyclohexylidene-protected diol (±)-13, instead favoring allylation of the 1-O-position and leaving the 6-O-position free for derivatization with a chiral resolving reagent and subsequent glycosylation. This hypothesis was based on the common observation that, in stannylene acetal-directed alkylations, an equatorial hydroxyl group in a 6-membered ring system reacts preferentially if neighbored by an axial substituent.42 Conversely, the bulky cyclohexylidene rings of compound (±)-13 distort its conformation, resulting in its atypical selectivity under identical reaction conditions.42 The predicted reversal of selectivity was realized in the event, and compound (±)-8 was obtained in 72% yield.
:1 ratio to give compound 14 in high yield (Scheme 2A). Translation of this strategy to a suitable PMB-protected inositol derivative was halted because we were unable to identify a common orthogonal protecting group (PG) for subsequent protection of the 1-O-position that satisfied the following criteria: (1) PG must not be susceptible to cleavage or migration by strong acid (AcCl/MeOH) or base (NaH/PMBCl); (2) PG must be orthogonal to PMB, allyl, tert-butyldimethylsilyl (TBS), and azido groups. Therefore, a new route based on delayed 1,6-O-differentiation was devised (Scheme 2B). The 1-O- and 6-O-positions of racemic diol (±)-13 were simultaneously protected as allyl ethers, which were unaffected by the following cyclohexylidene removal and para-methoxybenzylation reactions. Pd-catalyzed deallylation of bis-allyl ether (±)-17 was inefficient (yield < 50%), so alternative methods were explored. Cha's protocol, which employs Ti(Oi-Pr)4 and a commercially available Grignard reagent,41 cleanly removed both allyl groups to provide inositol (±)-18 in 85% yield. For the delayed 1,6-O-differentiation of PMB-protected diol (±)-18, we expected that the regioselectivity of stannylene acetal-directed alkylation would fortuitously invert with respect to cyclohexylidene-protected diol (±)-13, instead favoring allylation of the 1-O-position and leaving the 6-O-position free for derivatization with a chiral resolving reagent and subsequent glycosylation. This hypothesis was based on the common observation that, in stannylene acetal-directed alkylations, an equatorial hydroxyl group in a 6-membered ring system reacts preferentially if neighbored by an axial substituent.42 Conversely, the bulky cyclohexylidene rings of compound (±)-13 distort its conformation, resulting in its atypical selectivity under identical reaction conditions.42 The predicted reversal of selectivity was realized in the event, and compound (±)-8 was obtained in 72% yield.
 | ||
| Scheme 2 (A) Failed preparation of 8 using initial 1,6-O-differentiation. (B) Synthesis of 8via delayed 1,6-O-differentiation. (a) NaH, AllBr, DMF, 81%. (b) AcCl, CH2Cl2, MeOH, 98%. (c) NaH, PMBCl, DMF, 73%. (d) Ti(Oi-Pr)4, cyclohexylmagnesium chloride, THF, Et2O, 85%. (e) Bu2SnO, toluene, reflux; AllBr, CsF, DMF, 72%. (f) (1S)-(−)-camphanic chloride, DMAP, Et3N, CH2Cl2, 46%. (g) 1 M NaOH, MeOH, THF, 95%. | ||
Enantiomeric resolution of racemate (±)-8 was achieved by acylation of the free 6-O-position with chiral reagent (1S)-(−)-camphanic chloride to yield HPLC-separable diastereomers. This step also confirmed the regioselectivity of the preceding allylation reaction, as 1H NMR data showed that the inositol 6-H shifted downfield (δ 4.08 → 5.71), indicating that acylation occurred at the desired 6-O-position. Finally, diastereomer (−)-19 was subjected to saponification to provide optically pure inositol 8 in 44% yield over two steps (maximum yield 50%). The correct enantiomer was identified by comparison of optical rotation to an authentic sample synthesized by the same route starting from optically pure diol (+)-13. The chemical resolution of inositol described here (two steps, 44% yield) compares favorably to the frequently used enzymatic resolution reported by Gou and co-workers (3 steps, 20% yield),43 but the requirement for HPLC purification of (−)-19 could be problematic for large scale synthesis.
As coupling partners for 8, glucosaminyl donors 9 and 30, bearing trichloroacetimidate and fluoride as leaving groups, respectively, were synthesized through a series of similar transformations from 2-azido-1,3,4,6-tetra-O-acetyl-2-deoxy-D-glucopyranoside (20). En route to 9 (Scheme 3A), the poorly reactive anomeric acetate of 20 was unresponsive to treatment with AllOH/BF3·OEt2, but was cleanly displaced by AllOH in the presence of SnCl4 after stirring at room temperature for 72 h. To set up for discrimination of the 4-O-position, intermediate 21 was sequentially subjected to basic deacetylation and acid-catalyzed transacetalization with para-anisaldehyde dimethyl acetal to form 4,6-O-(para-methoxybenzylidene)-protected intermediate 22. After protection of the free 3-O-position with a PMB group to afford 23, the 4,6-O-(para-methoxybenzylidene) ring was regioselectively opened with dry HCl and NaBH3CN, exposing the 4-O-position as desired. This site was then protected with a TBS group to give 24, which proceeded efficiently with TBSOTf but failed with TBSCl, emphasizing the low reactivity of the 4-O-position. Next, deallylation of the anomeric position with PdCl2 did not generate the desired hemiacetal, but instead likely resulted in a 1,3-dipolar cycloaddition with the adjacent 2-azido group as previously reported.44 However, Ir-catalyzed isomerization to the corresponding vinyl ether and subsequent hydrolysis by Hg(II)45 produced hemiacetal 25 in 85% yield. Finally, conversion of the hemiacetal to glycosyl trichloroacetimidate 9 was accomplished using trichloroacetonitrile and 1,8-diazabicycloundec-7-ene (DBU). Glycosyl fluoride 27, which was obtained from 20via anomeric deacetylation and fluorination with diethylaminosulfur trifluoride (DAST), was elaborated to glycosyl fluoride donor 30 as described for compound 9 with comparable yields (Scheme 3B).
![Synthesis of donors 9 and 30. (a) AllOH, SnCl4, MS 4 Å, CH2Cl2, 81%. (b) NaOMe, MeOH. (c) anisaldehyde dimethyl acetal, CSA, DMF, 77% for 22, 78% for 28 (two steps). (d) NaH, PMBCl, DMF, 97% for 23, 95% for 29. (e) NaBH3CN, dry HCl in Et2O, MS 4 Å, THF. (f) TBSOTf, 2,6-lutidine, CH2Cl2, 60% for 24, 72% for 30 (two steps). (g) [Ir(COD)(PMePh2)2]PF6, H2, THF; then HgCl2, HgO, acetone, H2O, 85%. (h) Cl3CCN, DBU, CH2Cl2, 83%. (i) BnNH2, THF, 87%. (j) DAST, CH2Cl2, 98%.](/image/article/2011/SC/c1sc00440a/c1sc00440a-s3.gif) | ||
| Scheme 3 Synthesis of donors 9 and 30. (a) AllOH, SnCl4, MS 4 Å, CH2Cl2, 81%. (b) NaOMe, MeOH. (c) anisaldehyde dimethyl acetal, CSA, DMF, 77% for 22, 78% for 28 (two steps). (d) NaH, PMBCl, DMF, 97% for 23, 95% for 29. (e) NaBH3CN, dry HCl in Et2O, MS 4 Å, THF. (f) TBSOTf, 2,6-lutidine, CH2Cl2, 60% for 24, 72% for 30 (two steps). (g) [Ir(COD)(PMePh2)2]PF6, H2, THF; then HgCl2, HgO, acetone, H2O, 85%. (h) Cl3CCN, DBU, CH2Cl2, 83%. (i) BnNH2, THF, 87%. (j) DAST, CH2Cl2, 98%. | ||
With inositol derivative 8 and glucosaminyl donors 9 and 30 in hand, the formation of pseudodisaccharide 31 was investigated (Table 1). We first evaluated the reactions of glycosyl fluoride 30 (entries 1–3) because analogous benzyl-protected fluoride donors required only mild activation conditions for successful coupling to inositol in previous GPI syntheses.46,47 Unfortunately, the PMB-protected glycosyl fluoride was quite unreactive (entry 1). As shown, when performing this reaction with donor 30 at room temperature using any activation reagent and stirring for days, either no reaction or only trace amounts of the desired product were observed. Heating the reaction to reflux in Et2O completely consumed the donor, although only moderate yields of desired pseudodisaccharide 31 were obtained with Cp2HfCl2/AgOTf48 (22%) or Yb(OTf)3/CaCO349 (40%) as promoters (entries 2–3). The remainder of the mass balance was attributed to a side reaction resulting from the increased temperature required for donor activation (Fig. 3). It is proposed that upon heating donor 30 underwent ring flip and then activation, generating an oxocarbenium cation intermediate that possessed the proper conformation to undergo formation of 1,6-anhydro sugar 32, which was isolated and characterized. This product resulted from cleavage of the 6-O-PMB group and attack of the liberated primary alcohol onto the nearby electrophilic anomeric center. This was accompanied by trapping of the electrophilic PMB species by inositol acceptor 8, resulting in the formation of fully-protected inositol derivative 33, which was identified by MALDI MS.
|
|
|||
|---|---|---|---|
| Entry | Donor (X) | Conditions | Yield (α:β) |
| 1 | 30 (F) | Any activator/solvent, MS 4 Å, rt, days | No reaction |
| 2 | 30 (F) | Cp2HfCl2, AgOTf, MS 4 Å, Et2O, reflux, overnight | 22% (1:1) |
| 3 | 30 (F) | Yb(OTf)3, CaCO3, MS 4 Å, Et2O, reflux, overnight | 40% (2:1) |
| 4 | 9 (OC(NH)CCl3) | TMSOTf (cat.), MS 4 Å, CH2Cl2, 0 °C, 10 min | 84% (1:1.6) |
| 5 | 9 (OC(NH)CCl3) | TMSOTf (cat.), MS 4 Å, Et2O, 0 °C, 10 min | 86% (1.2:1) |
| 6 | 9 (OC(NH)CCl3) | TMSOTf (cat.), MS 4 Å, toluene-1,4-dioxane (1:1), 0 °C, 10 min |
80% (2.3:1) |

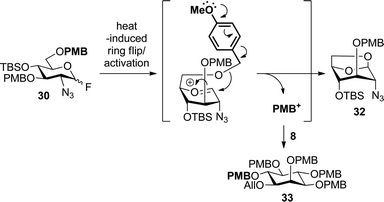 | ||
| Fig. 3 Possible mechanism of 1,6-anhydro sugar formation. | ||
Given the ineffectiveness of the glycosyl fluoride donor under numerous conditions, attention was therefore focused on the trichloroacetimidate donor (Table 1, entries 4–6). We expected that the low-temperature activation of trichloroacetimidate donors would eliminate the side reaction discussed above. Fortunately, when trichloroacetimidate 9 was employed, the desired pseudodisaccharide 31 was formed in 84% yield under standard Schmidt glycosylation conditions50 using catalytic TMSOTf at 0 °C in CH2Cl2. However, the undesired β-anomer was favored (α:β = 1:1.6), even when reported α-enhancing additives such as thiophene or phenylthioethyl ether51 were used. The anomeric ratio was somewhat improved by changing the solvent to Et2O, which resulted in an 86% yield and generated the α-anomer in slight excess (α:β = 1.2:1), whereas using toluene-1,4-dioxane (1:1) as the solvent52 resulted in an 80% yield and substantially increased α-stereoselectivity (α:β = 2.3:1). The anomers were inseparable by flash chromatography but could be purified by HPLC, or more conveniently by flash chromatography following deallylation, which gave 6 in 96% yield when using the aforementioned Ir–Hg protocol.
The resulting intermediate 6, bearing a free hydroxyl group at the inositol 1-O-position, was then phospholipidated using a phosphoramidite53 with the desired lipid chains (Scheme 4). For target GPIs 1–5, we opted to install a distearoyl phospholipid, which was accomplished using freshly prepared phosphoramidite 34 in the presence of 1H-tetrazole. Oxidation of the intermediate phosphite to a phosphate was performed in situ using t-BuOOH at 0 °C, which generated a 1:1 mixture of diastereomers originating from the stereogenic phosphorus atom. Subsequent removal of the glucosamine 4-O-TBS group proved difficult, which was consistent with the low accessibility of this position observed during TBS installation. The intermediate was unreactive toward tetrabutylammonium fluoride (TBAF) buffered with acetic acid. When stronger conditions were used, the starting material decomposed. For example, unbuffered TBAF cleaved the base-labile cyanoethoxyl phosphate protecting group prior to cleaving the TBS group. More acidic reagents, such as pyridine·HF, resulted in hydrolysis of multiple PMB groups prior to affecting the TBS group. Finally, we found that triethylamine trihydrofluoride cleanly effected the desired desilylation without compromising other protecting groups, although reaction times were quite long (5–7 days). The desired pseudodisaccharide 35 was obtained in 54% yield over two steps. Separation of this 1:1 diastereomeric mixture by HPLC was carried out to facilitate characterization of downstream GPI intermediates by NMR spectroscopy.
 | ||
| Scheme 4 Synthesis of pseudodisaccharide 35. (a) 1H-tetrazole, CH3CN, CH2Cl2; then t-BuOOH, 0 °C. (b) Et3N·3HF, THF, CH3CN, 54% (two steps). | ||
Synthesis of trimannoside 7
Trimannose glycoside 7 was designed to contain orthogonal protecting groups at the Man-I 2-O-position and Man-III 6-O-position for late-stage functionalization and phosphorylation, respectively, while the Man-I anomeric thioether moiety was chosen due to its high adaptability as a leaving group in glycosylation reactions. The preparation of trimannoside 7 involved the α-stereoselective coupling of mannose building blocks 10–12, which were prepared from a common intermediate, mannose pentaacetate 36 (Scheme 5). The Man-I building block, thioglycoside 10, was generated from known 4,6-O-(para-methoxybenzylidene) diol 3754 in three steps. To differentiate the 2-O- and 3-O-positions, a PMB group was installed at the latter position via a stannylene acetal-directed regioselective alkylation. Next, the exposed 2-O-position, which was marked for later functionalization, was masked with an orthogonal allyl group by treatment with NaH and AllBr, generating compound 39 in 68% yield over two steps. Then, the 4,6-O-(para-methoxybenzylidene) ring was regioselectively opened using diisobutylaluminium hydride (DIBAL-H), which exposed the 6-O-position hydroxyl group and provided compound 10 in 82% yield. Man-II glycosyl donor 11 was prepared from 36via orthoester 40,55 which underwent NaOMe-catalyzed deacetylation and para-methoxybenzylation. This intermediate was treated with acetic acid to regioselectively open the orthoester ring, providing a 2-O-acetyl hemiacetal that was converted to the corresponding trichloroacetimidate 11 by treatment with trichloroacetonitrile and DBU. Synthesis of Man-III donor 12 started with a BF3-promoted glycosylation of 36 with allyl alcohol. Subsequent deacetylation and selective protection of the 6-O-position by reaction with para-methoxytrityl (PMTrt) chloride afforded triol 41. Then, para-methoxybenzylation of the 2-, 3-, and 4-O-positions was followed by exchanging the PMTrt group for a TBS group to give 42. This protecting group manipulation was necessary because NaH-promoted para-methoxybenzylation in the presence of a 6-O-TBS group resulted in silyl ether migration. Finally, Pd-catalyzed deallylation revealed the anomeric hydroxyl group, which was transformed into trichloroacetimidate 12via treatment with trichloroacetonitrile and DBU.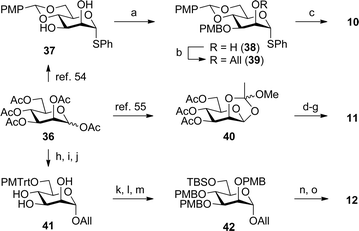 | ||
| Scheme 5 Synthesis of mannose building blocks 10–12. (a) Bu2SnO, toluene, reflux; PMBCl, CsF, DMF. (b) NaH, AllBr, DMF, 68% (two steps). (c) DIBAL-H, CH2Cl2, 82%. (d) NaOMe, MeOH. (e) NaH, PMBCl, DMF, 76% (two steps). (f) AcOH–H2O (1:1), 70%. (g) Cl3CCN, DBU, CH2Cl2, 77%. (h) AllOH, BF3·OEt2, MS 4 Å, CH2Cl2, 66% (two steps from D-mannose). (i) NaOMe, MeOH. (j) PMTrtCl, pyridine, 66% (two steps). (k) NaH, PMBCl, DMF, 73%. (l) AcOH, H2O, CH2Cl2, 94%. (m) TBSCl, pyridine, 88%. (n) PdCl2, AcOH, NaOAc, CH2Cl2, H2O, 82%. (o) Cl3CCN, DBU, CH2Cl2, 73%. | ||
With monosaccharides 10–12 in hand, the construction of trimannoside 7 commenced (Scheme 6). Schmidt conditions were employed for both glycosylations based on the successful results obtained in the inositol-glucosamine coupling. The glycosylation of Man-I acceptor 10 by Man-II trichloroacetimidate donor 11 proceeded smoothly using catalytic TMSOTf in CH2Cl2 at 0 °C. The participating 2-O-acetyl group of 11 ensured generation of only α-product 43. Deacetylation with K2CO3/MeOH exposed the 2-O-position, resulting in dimannoside alcohol 44 in 66% yield over two steps. Coupling of 44 with Man-III trichloroacetimidate 12 under the same conditions gave trimannoside 7 in a 5:1 α:β ratio, a figure that was improved to α-only (76% yield) upon switching the solvent to Et2O. The stereochemical configurations of all three anomeric centers were established as α based on their anomeric 13C NMR JCH coupling constants (Man-I-JCH = 167 Hz, Man-II-JCH = 171 Hz, Man-III-JCH = 171 Hz).56 With the two key glycosylation components 6 and 7 ready, we proceeded to the construction of the target GPI anchors.
 | ||
| Scheme 6 Synthesis of trimannoside 7. (a) TMSOTf (cat.), MS 4 Å, CH2Cl2, 0 °C. (b) K2CO3, MeOH, 66% (two steps). (c) 12, TMSOTf (cat.), MS 4 Å, Et2O, 0 °C, 76%. | ||
Synthesis of first generation clickable GPI 1
The strategy used for the synthesis of first generation Alkynyl-GPI 1 was based on the following sequence: (1) convergent coupling of the pseudodisaccharide and trimannose fragments; (2) phosphorylation of the Man-III 6-O-position; (3) late-stage acylation of the Man-I 2-O-position to install the alkyne click tag; (4) global deprotection. We first attempted the key glycosylation reaction by direct activation of thioglycoside donor 7 with reagents that would not affect the allyl group. Reagents in this category, such as dimethyl(methylthio)sulfonium trifluoromethanesulfonate (DMTST)57 and para-toluenesulfenyl chloride (p-TolSCl)/AgOTf58 in conjunction with hindered base 2,4,6-tri-tert-butylpyrimidine (TTBP),59 delivered the desired compound, albeit in low yields (< 25%). As shown in Scheme 7, the glycosylation was significantly improved by employing the corresponding trichloroacetimidate donor 45, which was produced from compound 7 in two steps, including hydrolysis of the anomeric thioether by p-TolSCl/AgOTf/TTBP to give the hemiacetal and subsequent treatment with trichloroacetonitrile and DBU. Activation of 45 by catalytic TMSOTf in the presence of pseudodisaccharide acceptor 35 generated the desired GPI intermediate, which after work up was reacted with triethylamine trihydrofluoride to cleanly remove the Man-III 6-O-TBS group. The resulting pseudopentasaccharide alcohol 46 was obtained in 83% yield over two steps and fully characterized by high resolution mass spectrometry and NMR spectroscopy. The stereochemical configurations of all mannosidic bonds were established as α (anomeric 13C NMR JCH coupling constants for mannose units: JCH = 167 Hz, JCH = 174 Hz, JCH = 174 Hz).56 The glycosylation was quite clean, as no anydro sugar products or β-anomer were isolated on purification. Subsequently, a phosphoethanolamine group was introduced to the Man-III 6-O-position, for which phosphoramidite 47 was used in conjunction with promoter 1H-tetrazole. The reaction proceeded smoothly, forming the trivalent phosphite intermediate in 1 h. After cooling the reaction mixture to −40 °C, t-BuOOH was added to chemoselectively oxidize the phosphite to phosphate 48in situ without affecting the Man-I 2-O-allyl group. The phosphorylation proceeded in 81% yield and, as previously, generated a 1:1 mixture of diastereomers that was separated by HPLC to facilitate characterization. Next, removal of the Man-I 2-O-allyl group of 48 was accomplished by using the Ir–Hg protocol, which afforded secondary alcohol 49 in 90% yield.
![Key glycosylation and phosphorylation reactions. (a) p-TolSCl, AgOTf, TTBP, wet CH2Cl2, 50% (68% BRSM). (b) Cl3CCN, DBU, CH2Cl2, 92%. (c) 35, TMSOTf (cat.), MS 4 Å, Et2O. (d) Et3N·3HF, THF, CH3CN, 83% (two steps). (e) 47, 1H-tetrazole, CH2Cl2, CH3CN; then t-BuOOH, −40 °C, 81% (two steps). (f) [Ir(COD)(PMePh2)2]PF6, H2, THF; then HgCl2, HgO, acetone, H2O, 90%.](/image/article/2011/SC/c1sc00440a/c1sc00440a-s7.gif) | ||
| Scheme 7 Key glycosylation and phosphorylation reactions. (a) p-TolSCl, AgOTf, TTBP, wet CH2Cl2, 50% (68% BRSM). (b) Cl3CCN, DBU, CH2Cl2, 92%. (c) 35, TMSOTf (cat.), MS 4 Å, Et2O. (d) Et3N·3HF, THF, CH3CN, 83% (two steps). (e) 47, 1H-tetrazole, CH2Cl2, CH3CN; then t-BuOOH, −40 °C, 81% (two steps). (f) [Ir(COD)(PMePh2)2]PF6, H2, THF; then HgCl2, HgO, acetone, H2O, 90%. | ||
With the Man-I 2-O-position exposed in 49, the compound was ready for attachment of the click tag (Scheme 8). We first attempted direct esterification of the secondary hydroxyl group of intermediate 49 using alkyne-containing carboxylic acids under standard coupling conditions, e.g. dicyclohexyl carbodiimide (DCC) and DMAP. However, even at elevated temperatures, the desired acylation products were not formed, and starting material was recovered quantitatively. Other esterification methods for direct installation of the alkyne group also failed. However, it was discovered that the substrate could undergo succinylation in 67% yield when employing a large excess of succinic anhydride, DMAP, and MS 4 Å. Addition of a succinate linker to the GPI provided a carboxylic acid handle in 50, thus allowing for a much more facile amidation reaction to attach the alkyne. Accordingly, GPI carboxylic acid 50 was treated with propargyl amine under standard amide bond-forming conditions, namely, employing 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) and 1-hydroxybenzotriazole (HOBt). This reaction successfully installed the click tag, furnishing fully protected alkyne-modified GPI 51 in 75% yield.
 | ||
| Scheme 8 Synthesis of first generation Alkynyl-GPI 1. (a) excess succinic anhydride, DMAP, CH2Cl2, MS 4 Å, 67%. (b) propargyl amine, EDCI, HOBt, CH2Cl2, DMF, 75%. (c) Zn, AcOH, CH2Cl2, 1 h; DBU, CH2Cl2 1 h; CH2Cl2–TFA (9:1), 30 min, 85% (0.40 mg, three steps). | ||
The final phase of synthesis involved the development of a suitable global deprotection procedure, which required three sets of reaction conditions: (1) reductive, to convert the azidoglucose moiety to glucosamine; (2) basic, to remove the Fmoc and cyanoethoxyl protecting groups; (3) acidic or oxidative, to remove the global PMB protecting groups. After optimization of reaction concentrations, times, and sequence, a three-step, one-pot reaction was developed that could efficiently remove all of the protecting groups in less than 3 h: (1) zinc-mediated reduction of the azide; (2) after filtration and evaporation, removal of the base-labile Fmoc and cyanoethoxyl groups by treatment with DBU; (3) direct addition of a TFA solution to a final concentration of 10%, which quenched the DBU and cleanly removed all PMB groups in only 30 min. This protocol was applied to the global deprotection of 51, and subsequent purification of the target GPI was accomplished using a Sephadex LH-20 size exclusion column employing CHCl3–CH3OH–H2O (3:3:1) as the mobile phase to obtain Alkynyl-GPI 1. Although compound 1 was initially isolated and characterized in pure form in 85% yield (0.40 mg), we observed cleavage of the Man-I 2-O-acyl group after several weeks of storage at 4 °C. This decomposition did not occur during the global deprotection procedure, because all reactions monitored by TLC and MALDI MS showed clean conversion at each step. We speculate that decomposition may have resulted from an intramolecular cleavage mechanism that is similar to aspartimide formation in solid-phase peptide synthesis.60 With our attention raised to the unexpected linker instability of GPI 1, we set out to construct a series of more stable GPIs whose functional tags would remain intact during chemical synthesis and biological applications.
Synthesis of second generation clickable Alkynyl-GPI 2
To circumvent the disappointing decomposition of the acyl-linked click tag in GPI 1, we set out to synthesize the second generation Alkynyl-GPI 2 containing a stable alkyl-based linker at the Man-I 2-O-position (Scheme 9). As late-stage alkylation would typically require harsh conditions (e.g., using NaH) that would compromise protecting groups in a fully elaborated GPI intermediate, the alkyne click tag was attached to the trimannose fragment prior to the key glycosylation reaction. An additional benefit of this strategy was the elimination of risky late-stage manipulations to install the functional tag. Thus, we targeted alkyne-modified trimannoside 55 as the glycosyl donor. Starting from intermediate thioglycoside 7, we explored conditions for the removal of the Man-I 2-O-allyl group. When traditional Pd-based methods failed, likely due to catalyst poisoning by the anomeric thioether group, we again turned to Cha's Ti(IV)-mediated deallylation protocol, which delivered the desired compound 52 in 82% yield. Treatment of this intermediate with NaH and propargyl bromide installed an alkyne group at the Man-I 2-O-position, affording 53 in 78% yield. Subsequent hydrolysis of the anomeric thioether was carried out in 77% yield using para-nitrobenzenesulfenyl chloride/AgOTf/TTBP61 in lieu of N-iodosuccinimide (NIS)/AgOTf so as not to affect the alkyne. The resulting hemiacetal 54 was treated with trichloroacetonitrile and DBU to provide donor 55 in 90% yield. | ||
| Scheme 9 Synthesis of second generation Alkynyl-GPI 2. (a) Ti(Oi-Pr)4, cyclopentylmagnesium chloride, THF, Et2O, 82%. (b) NaH, propargyl bromide, DMF, 78%. (c) para-nitrobenzenesulfenyl chloride, AgOTf, TTBP, wet CH2Cl2, 77%. (d) Cl3CCN, DBU, CH2Cl2, 90%. (e) 35, TMSOTf (cat.), MS 4 Å, Et2O. (f) Et3N·3HF, THF, CH3CN, 82% (two steps). (g) 47, 1H-tetrazole, CH2Cl2, CH3CN; then t-BuOOH, −40 °C, 72% (two steps). (h) Zn, AcOH, CH2Cl2, 1 h; DBU, CH2Cl2 1 h; CH2Cl2–TFA (9:1), 30 min, 93% (1.03 mg, three steps). | ||
The coupling of 2-O-propargyl-functionalized donor 55 with acceptor 35 using catalytic TMSOTf in Et2O delivered the expected GPI intermediate. After brief work up but without purification, the intermediate was treated with triethylamine trihydrofluoride to remove the Man-III 6-O-TBS group, providing exclusively alkyne-modified α-pseudopentasaccharide 56 in 82% yield over two steps. This intermediate was fully characterized using high resolution mass spectrometry and NMR spectroscopy, and the stereochemical configurations of all mannosidic bonds were established as α (anomeric 13C NMR JCH coupling constants for mannose units: JCH = 171 Hz, JCH = 172 Hz, JCH = 177 Hz).56 Phosphorylation of the exposed Man-III 6- O-position of compound 56 was performed as described above, employing phosphoramidite 47 and 1H-tetrazole followed by chemoselective in situ oxidation by t-BuOOH at −40 °C. The resulting fully protected GPI intermediate 57 was obtained in 72% yield as an inconsequential 1:1 mixture of diastereomers.
Finally, compound 57 was subjected to the established three-step, one-pot global deprotection, which entailed sequential treatment with Zn/AcOH, DBU, and 10% TFA. The deprotection was accomplished in under 3 h, and purification of the reaction mixture by Sephadex LH-20 size exclusion chromatography provided Alkynyl-GPI 2 in 93% yield (1.03 mg). MALDI MS and 1H/31P NMR spectroscopy were used to characterize and confirm the homogeneity of the target compound. The alkyne tag of GPI 2 was unaffected by the global deprotection conditions and the alkyl-based linker proved stable during storage.
Synthesis of second generation clickable Azido-GPI 3
While the alkyne group has enjoyed great success as a tag for click chemistry, the azide has been touted as a superior click handle, in part because it can undergo Cu-free strain-promoted [3 + 2] cycloadditions with cyclooctyne-based probes.62 To avoid potentially toxic effects of Cu(I), bioorthogonal chemistries such as the Staudinger ligation63 and azide-cyclooctyne [3 + 2] cycloaddition are desirable for in vivo studies. Therefore, we set out to prepare Azido-GPI 3 to further expand our toolbox of functionalized GPIs.Synthetically, the presence of an intact azido group in the target GPI is problematic for existing strategies because the glucosamine moiety commonly contains an azide, which not only acts as a latent amino group but also permits α-stereoselectivity in the inositol glycosylation. To address this issue, we decided to exchange the azide for an Fmoc-protected amino group on intermediate 6 (Scheme 10), which had already exploited the non-participating property of the 2-azido group to stereoselectively forge the glucosamine-inositol bond. Thus, compound 6 was subjected to a one-pot procedure that employed Zn-mediated azide reduction followed by treatment with excess FmocCl and NaHCO3, which was performed in the presence of water to prevent reaction at the free inositol 1-O-position. The resulting compound 58, isolated in 78% yield, underwent phosphorylation of the inositol 1-O-position using phospholipid precursor 34 in the presence of 1H-tetrazole. After in situ oxidation by t-BuOOH and work up, intermediate phosphate 59 was treated with triethylamine trihydrofluoride to cleave the glucosamine 4-O-TBS group, which gave pseudodisaccharide 60 in 67% yield over two steps. To simplify characterization of 60 and its downstream intermediates, the 1:1 diastereomeric mixture resulting from phosphorylation was separated by HPLC.
 | ||
| Scheme 10 Synthesis of modified pseudodisaccharide 60. (a) Zn, AcOH, CH2Cl2. (b) FmocCl, NaHCO3, 1,4-dioxane–H2O (10:1), 78% (two steps). (c) 34, 1H-tetrazole, CH2Cl2, CH3CN; then t-BuOOH, 0 °C. (d) Et3N·3HF, THF, CH3CN, 67% (two steps). | ||
With Fmoc-protected acceptor 60 in hand, its coupling partner trimannose donor 63 was synthesized (Scheme 11). As in the construction of Alkynyl-GPI 2, we elected to install the azido group early to eliminate late-stage manipulations. Thus, azide-modified trimannose 62 was generated from its secondary alcohol precursor 52via a four step sequence64 including alkylation of the Man-I 2-O-position with tert-butyl bromoacetate, reduction of the newly formed tert-butyl ester using LiAlH4 (59% over two steps), and mesylation of the resulting primary hydroxyl group followed by displacement with sodium azide at 90 °C to produce compound 62 (82% over two steps). Donor 63 was prepared by hydrolysis of the anomeric thioether with NIS/AgOTf/TTBP in wet CH2Cl2 followed by treatment of the resulting hemiacetal with trichloroacetonitrile and DBU.
 | ||
| Scheme 11 Synthesis of second generation Azido-GPI 3. (a) NaH, tert-butyl bromoacetate, DMF, 77%. (b) LiAlH4, THF, 77%. (c) MsCl, DIPEA, CH2Cl2. (d) NaN3, DMF, 90 °C, 82% (two steps). (e) NIS, AgOTf, TTBP, wet CH2Cl2, 71%. (f) Cl3CCN, DBU, CH2Cl2, 96%. (g) 60, TMSOTf (cat.), MS 4 Å, CH2Cl2. (h) Et3N·3HF, THF, CH3CN, 30% (two steps, 88% BRSM). (i) 47, 1H-tetrazole, CH2Cl2, CH3CN; then t-BuOOH, 0 °C, 61% (two steps). (j) DBU, CH2Cl2 1 h; CH2Cl2–TFA (9:1), 30 min, 97% (0.76 mg, two steps). | ||
In the key glycosylation reaction, acceptor 60 and donor 63 combined in the presence of catalytic TMSOTf at 0 °C in CH2Cl2. After work up, the crude mixture was subjected to desilylation with triethylamine trihydrofluoride. On purification, the desired α-pseudopentasaccharide 64 was obtained in a moderate 30% yield over two steps (88% based on recovery of acceptor 60). GPI intermediate 64 was fully characterized using MALDI MS and NMR spectroscopy, including the stereochemical configurations of all mannosidic bonds, which were established as α (anomeric JCH coupling constants for mannose units: JCH = 166 Hz, JCH = 174 Hz, JCH = 175 Hz).56 Phosphorylation of the Man-III 6-O- position, conducted according to the procedure described for 56 (Scheme 9), provided the desired intermediate as an inconsequential 1:1 mixture of diastereomers in 61% isolated yield. Because azide reduction was unnecessary in the final sequence, the global deprotection was simplified to a two-step, one-pot procedure requiring only treatment with DBU followed by 10% TFA, which cleanly removed all protecting groups in under 2 h. Purification of the reaction mixture by Sephadex LH-20 size exclusion chromatography provided Azido-GPI 3 in 97% yield (0.76 mg), and its structure and homogeneity were confirmed by MALDI MS and 1H/31P NMR spectroscopy. The azide tag was unaffected by the global deprotection conditions and the alkyl-based linker was stable during storage.
Although the clickable GPIs 2 and 3 were each obtained in a relatively small quantity of ∼1 mg, we expect that their syntheses will be amenable to scale-up, particularly because the second generation modified routes obviated the need for late-stage manipulations to install the click tag. The primary point for improvement will be to limit HPLC purification. In the present work, HPLC was used to separate inconsequential diastereomeric mixtures to simplify characterization, which will not be necessary during scale-up. HPLC was also used in the synthesis of inositol 8, although this may be mitigated by using enzymatic resolution43 and exploring alternative routes such as de novo synthesis of PMB-protected inositols.65 Finally, the moderate stereoselectivity obtained in the formation of pseudodisaccharide 31, while consistent with our previous experience, may be improved by investigating recently published methods of stereoselective 1,2-cis-2-amino glycoside formation such as Nguyen's Ni-catalyzed glycosylation.66 Overall, PMB protection is comparable to benzyl protection in terms of efficiency, while offering an immense improvement in functional group tolerance. We expect to be able to access appreciable quantities (10–100 mg range) of clickable GPI anchors on scale-up. Furthermore, the flexibility imparted by our convergent–divergent strategy will allow us to alter the functionalization site in the trimannose moiety if necessary.
Click reactions of GPIs 2 and 3 to generate GPI-Fluor 4 and GPI-Biotin 5
To validate fully deprotected Alkynyl-GPI 2 and Azido-GPI 3 as clickable tools for the biological study of GPI anchors, we explored their coupling reactions with imaging and affinity probes (Scheme 12). Alkynyl-GPI 2 was conjugated with Azide-Fluor 488 (65), commercially available as a regioisomeric mixture, via Cu(I)-catalyzed [3 + 2] cycloaddition under standard conditions.67 The reaction was monitored by MALDI MS, which showed complete conversion of the starting material to GPI-Fluor conjugate 4, a promising tool for visualizing GPI behavior in cellular contexts. Azido-GPI 3 was effectively coupled via Cu-free strain-promoted [3 + 2] cycloaddition to BARAC-biotin (66), a biarylazacyclooctynone-based affinity probe developed by Jewett et al.68 MALDI MS confirmed virtually quantitative conversion of the starting material to GPI-Biotin 5. Conjugation of GPIs to affinity tags such as biotin is a potentially useful strategy for pull-down experiments to probe cell surface GPI-anchored proteomics. GPIs 2 and 3, here demonstrated to undergo efficient coupling reactions via click chemistry, represent versatile tools for the development of chemical approaches to studying the biology of GPI anchors. | ||
| Scheme 12 Click reactions of GPIs 2 and 3 to form GPI-conjugates 4 and 5. (a) 5 equivalents Azide-Fluor 488 (65), CuSO4, sodium ascorbate, 37 °C, THF–MeOH–H2O (1:1:1). (b) BARAC-biotin (66), CHCl3–MeOH–H2O (3:3:1). | ||
Conclusions
GPI anchor research has advanced steadily over the past three decades, but the development of new tools that take advantage of emerging technologies is imperative to accelerate progress in the field. Given the rapid influx of techniques in biochemistry and chemical biology that use click chemistry for chemoselective conjugation, particularly the powerful [3 + 2] cycloaddition between azides and alkynes/cyclooctynes, arming GPIs with click tags provides a platform for developing novel approaches to exploring their biological properties and therapeutic applications. A challenging yet necessary approach to accessing useful quantities of homogeneous GPIs and GPI analogs is chemical synthesis, a task whose difficulty is compounded by the presence of certain classes of functional groups in synthetic targets. Click chemistry tags and other useful functionalities, including many imaging and affinity tags, are often incompatible with traditional protecting group chemistries used in carbohydrate synthesis. To address this shortcoming, we developed a strategy based on using the PMB group for permanent hydroxyl protection, which provides unprecedented flexibility for functional group incorporation in GPI synthesis.In this work, PMB protection was used in concert with a convergent–divergent strategy to synthesize functionalized GPIs 1–5 from two common intermediates, pseudodisaccharide 6 and trimannose thioglycoside 7. Overall, the preparation of PMB-protected monosaccharide and inositol building blocks was carried out efficiently. The performance of these intermediates in Schmidt glycosylation reactions was excellent, although in two cases we did observe de-para-methoxybenzylative anhydro sugar formation when attempting coupling of certain donors with poor glycosyl acceptors. After circumventing these obstacles, first generation target Alkynyl-GPI 1 was synthesized, but its acyl-based linker decomposed during storage. Therefore, second generation targets Alkynyl-GPI 2 and Azido-GPI 3, bearing stable alkyl-based linkers, were successfully synthesized using a modified synthetic strategy involving early functional tag installation. For global deprotection, we developed a two- or three-step, one-pot reaction that was performed in less than 3 h and afforded the target GPIs in excellent yields, ranging from 85 to 97%. Fully deprotected Alkynyl-GPI 2 and Azido-GPI 3 were chemoselectively labeled with fluorescent and affinity probes using Cu(I)-catalyzed and strain-promoted click reactions to give GPI-Fluor 4 and GPI-Biotin 5, respectively. These results demonstrate that compounds 2 and 3 should undergo efficient temporally controlled labeling via click chemistry in biological settings. The functionalized GPI anchors synthesized here, particularly clickable GPIs 2 and 3, offer a versatile toolset with which to study the process of GPI anchorage. Furthermore, the PMB protection strategy represents a general approach to the synthesis of functionalized GPIs and potentially other complex functionalized glycoconjugates.
Acknowledgements
This work was funded by NIH/NIGMS (R01GM090270). We thank Dr B. Shay and Dr L. Hryhorczuk for MS measurements, and Dr B. Ksebati for help with some NMR experiments. Dr S. Burgula is acknowledged for performing the click reaction between Alkynyl-GPI 2 and Azide-Fluor 488. We are grateful to Prof. X. Huang (Michigan State University) for providing p-TolSCl and Dr J. Jewett (Bertozzi group, University of California, Berkeley) for generously providing BARAC-biotin.References
- M. A. J. Ferguson and A. F. Williams, Annu. Rev. Biochem., 1988, 57, 121–138 CrossRef.
- P. T. Englund, Annu. Rev. Biochem., 1993, 62, 121–138 CrossRef CAS.
- H. Ikezawa, Biol. Pharm. Bull., 2002, 25, 409–417 CAS.
- M. G. Paulick and C. R. Bertozzi, Biochemistry, 2008, 47, 6991–7000 CrossRef CAS.
- M. A. Ferguson, J. Cell. Sci., 1999, 112, 2799–2809 CAS.
- B. Eisenhaber, P. Bork and F. Eisenhaber, Protein Eng., Des. Sel., 2001, 14, 17–25 CrossRef CAS.
- H. T. He, J. Finne and C. Goridis, J. Cell Biol., 1987, 105, 2489–2500 CrossRef CAS.
- P. J. Robinson, M. Millrain, J. Antoniou, E. Simpson and A. L. Mellor, Nature, 1989, 342, 85–87 CrossRef CAS.
- D. D. Eardley and M. E. Koshland, Science, 1991, 251, 78–81 CAS.
- G. A. M. Cross, Annu. Rev. Immunol., 1990, 8, 83–110 CrossRef CAS.
- K. Hwa, Adv. Exp. Med. Biol., 2001, 491, 207–214 CrossRef CAS.
- M. A. J. Ferguson, S. W. Homans, R. A. Dwek and T. W. Rademacher, Science, 1988, 239, 753–759 CAS.
- M. Fujita and Y. Jigami, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 410–420 CrossRef CAS.
- Y. Varma and T. Hendrickson, ChemBioChem, 2010, 11, 623–636 CrossRef CAS.
- L. Schofield, M. C. Hewitt, K. Evans, M.-A. Siomos and P. H. Seeberger, Nature, 2002, 418, 785–789 CrossRef CAS.
- C. Murakata and T. Ogawa, Tetrahedron Lett., 1991, 32, 671–674 CrossRef CAS.
- C. Murakata and T. Ogawa, Carbohydr. Res., 1992, 235, 95–114 CrossRef CAS.
- T. G. Mayer, B. Kratzer and R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1994, 33, 2177–2181 CrossRef.
- A. S. Campbell and B. Fraser-Reid, J. Am. Chem. Soc., 1995, 117, 10387–10388 CrossRef CAS.
- D. K. Baeschlin, A. R. Chaperon, V. Charbonneau, L. G. Green, S. V. Ley, U. Lücking and E. Walther, Angew. Chem., Int. Ed., 1998, 37, 3423–3428 CrossRef CAS.
- D. Tailler, V. Ferrières, K. Pekari and R. R. Schmidt, Tetrahedron Lett., 1999, 40, 679–682 CrossRef CAS.
- D. K. Baeschlin, A. R. Chaperon, L. G. Green, M. G. Hahn, S. J. Ince and S. V. Ley, Chem.–Eur. J., 2000, 6, 172–186 CrossRef CAS.
- M. Martín-Lomas, N. Khiar, S. García, J.-L. Koessler, P. M. Nieto and T. W. Rademacher, Chem.–Eur. J., 2000, 6, 3608–3621 CrossRef.
- K. Ruda, J. Lindberg, P. J. Garegg, S. Oscarson and P. Konradsson, J. Am. Chem. Soc., 2000, 122, 11067–11072 CrossRef CAS.
- M. C. Hewitt, D. A. Snyder and P. H. Seeberger, J. Am. Chem. Soc., 2002, 124, 13434–13436 CrossRef CAS.
- J. Xue and Z. Guo, J. Am. Chem. Soc., 2003, 125, 16334–16339 CrossRef CAS.
- J. Lu, K. N. Jayaprakash and B. Fraser-Reid, Tetrahedron Lett., 2004, 45, 879–882 CrossRef CAS.
- N. Shao, J. Xue and Z. Guo, Angew. Chem., Int. Ed., 2004, 43, 1569–1573 CrossRef CAS.
- A. Ali, D. C. Gowda and R. A. Vishwakarma, Chem. Commun., 2005, 519–521 RSC.
- X. Liu, Y.-U. Kwon and P. H. Seeberger, J. Am. Chem. Soc., 2005, 127, 5004–5005 CrossRef CAS.
- M. Hederos and P. Konradsson, J. Am. Chem. Soc., 2006, 128, 3414–3419 CrossRef CAS.
- D. V. Yashunsky, V. S. Borodkin, M. A. J. Ferguson and A. V. Nikolaev, Angew. Chem., Int. Ed., 2006, 45, 468–474 CrossRef CAS.
- X. Wu and Z. Guo, Org. Lett., 2007, 9, 4311–4313 CrossRef CAS.
- C. Becker, X. Liu, D. Olschewski, R. Castelli, R. Seidel and P. Seeberger, Angew. Chem., Int. Ed., 2008, 47, 8215–8219 CrossRef CAS.
- B. M. Swarts and Z. Guo, J. Am. Chem. Soc., 2010, 132, 6648–6650 CrossRef CAS.
- M. G. Paulick, A. R. Wise, M. B. Forstner, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2007, 129, 11543–11550 CrossRef CAS.
- M. G. Paulick, M. B. Forstner, J. T. Groves and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20332–20337 CrossRef CAS.
- We initially attempted the key glycosylation with a Man-I 2-O-PMB-protected trimannosyl donor. However, instead of forming the desired pseudopentasaccharide, the donor underwent de-methoxybenzylation to form a 1,2-anhydro sugar derivative. See Fig. S1 in the ESI† for more details.
- Z. Guo and L. Bishop, Eur. J. Org. Chem., 2004, 3585–3596 CrossRef CAS.
- J. Xue and Z. Guo, Bioorg. Med. Chem. Lett., 2002, 12, 2015–2018 CrossRef CAS.
- J. Lee and J. K. Cha, Tetrahedron Lett., 1996, 37, 3663–3666 CrossRef CAS.
- T. B. Grindley and R. Thangarasa, Can. J. Chem., 1990, 68, 1007–1019 CrossRef CAS.
- D.-M. Gou, Y.-C. Liu and C.-S. Chen, Carbohydr. Res., 1992, 234, 51–64 CrossRef CAS.
- C. Lamberth and M. D. Bednarski, Tetrahedron Lett., 1991, 32, 7369–7372 CrossRef CAS.
- J. J. Oltvoort, C. A. A. Van Boeckel, J. H. De Koning and J. H. Van Boom, Synthesis, 1981, 305–308 CrossRef.
- C. Murakata and T. Ogawa, Tetrahedron Lett., 1990, 31, 2439–2442 CrossRef CAS.
- P. J. Garegg, P. Konradsson, S. Oscarson and K. Ruda, Tetrahedron, 1997, 53, 17727–17734 CrossRef CAS.
- T. Matsumoto, H. Maeta, K. Suzuki and l. G.-i. Tsuchihashi, Tetrahedron Lett., 1988, 29, 3567–3570 CrossRef CAS.
- S. Hosono, W.-S. Kim, H. Sasai and M. Shibasaki, J. Org. Chem., 1995, 60, 4–5 CrossRef CAS.
- R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed. Engl., 1980, 19, 731–732 CrossRef.
- J. Park, S. Kawatkar, J.-H. Kim and G.-J. Boons, Org. Lett., 2007, 9, 1959–1962 CrossRef CAS.
- A. Demchenko, T. Stauch and G.-J. Boons, Synlett, 1997, 818–820 CrossRef CAS.
- W. Bannwarth and A. Trzeciak, Helv. Chim. Acta, 1987, 70, 175–186 CrossRef CAS.
- D. Crich and A. Banerjee, J. Am. Chem. Soc., 2006, 128, 8078–8086 CrossRef CAS.
- P. J. Garegg and L. Maron, Acta Chem. Scand., Ser. B, 1979, 33b, 39–41 Search PubMed.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297 RSC.
- P. Fügedi and P. J. Garegg, Carbohydr. Res., 1986, 149, C9–C12 CrossRef.
- V. Martichonok and G. M. Whitesides, J. Org. Chem., 1996, 61, 1702–1706 CrossRef CAS.
- D. Crich, M. Smith, Q. Yao and J. Picione, Synthesis, 2001, 323–326 CrossRef CAS.
- J. P. Tam, M. W. Riemen and R. B. Merrifield, Peptide Res., 1988, 1, 6–18 CAS.
- D. Crich, F. Cai and F. Yang, Carbohydr. Res., 2008, 343, 1858–1862 CrossRef CAS.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- S. Hagihara, A. Miyazaki, I. Matsuo, A. Tatami, T. Suzuki and Y. Ito, Glycobiology, 2007, 17, 1070–1076 CrossRef CAS.
- S. L. Bender and R. J. Budhu, J. Am. Chem. Soc., 1991, 113, 9883–9885 CrossRef CAS.
- E. A. Mensah, F. Yu and H. M. Nguyen, J. Am. Chem. Soc., 2010, 132, 14288–14302 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1, experimental details for the preparation of new compounds, MS and NMR spectra of new compounds. See DOI: 10.1039/c1sc00440a |
| This journal is © The Royal Society of Chemistry 2011 |