Photo-induced charge recombination kinetics in low bandgap PCPDTBT polymer:CdSe quantum dot bulk heterojunction solar cells†
Josep
Albero
a,
Yunfei
Zhou
b,
Michael
Eck
b,
Frank
Rauscher
d,
Phenwisa
Niyamakom
d,
Ines
Dumsch
e,
Sybille
Allard
e,
Ullrich
Scherf
e,
Michael
Krüger
b and
Emilio
Palomares
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), Avda. Països Catalans 16, E-43007, Tarragona, Spain. E-mail: epalomares@iciq.es; Fax: (+34) 977 920 224; Tel: (+34) 977920200
bFreiburg Materials Research Centre (FMF), University of Freiburg, Stefan Meier Strasse 21, D-79104, Freiburg, Germany. E-mail: Michael.krueger@fmf.uni-freiburg.de; Fax: (+49) (0)761 203 4701; Tel: (+49) (0)761 203 4755
cCatalan Institution for Research and Advanced Studies (ICREA), Avda. Lluis Companys 23, E-08010, Barcelona, Spain
dBayer Technology Services GmbH, Process Technology Building E41, D-51368, Leverkusen, Germany. Fax: (+49) 214 30 50262; Tel: (+49) 214 30 41798
eInstitut für Polymertechnologie, Bergishe Universität Wuppertal, Gauss-Strasse 20, D-42097, Wuppertal, Germany. E-mail: scherf@uni-wuppertal.de; Fax: (+49) (0)202 439 3880; Tel: (+49) (0)202 439 2493
First published on 20th September 2011
Abstract
The interfacial charge transfer recombination processes under working conditions that limit the device performance in polymer:CdSe quantum dot bulk heterojunction hybrid solar cells have been measured. The recombination lifetimes for electrons and holes in the device show an exponential dependence in a similar way to that observed for other molecular based solar cells such as bulk heterojunction organic solar cells (OSC) and dye sensitized solar cells (DSSC). The implications of this unprecedented observation on the design of novel devices are discussed as well as the relationship between the charge accumulation in these devices under operation and the device open-circuit voltage.
Introduction
Molecular based photovoltaic devices have received much attention in recent years due to their potential low cost and their increase in light-to-energy conversion efficiency. Metal oxide sensitized solar cells or Grätzel cells have obtained outstanding efficiencies under 1 sun (100 mW cm−2 sun simulated light) close to 11.5% and their stability has already been demonstrated for more than 1000 h.1,2 On the other hand, semiconductor polymers, in combination with excellent electron acceptors moieties such as fullerene derivates, have also received massive attention due to their application in solution processed organic solar cells with efficiencies close to 8.5%.3,4Indeed, the preliminary breakthrough stems from the widely known poly(3-hexylthiophene) (P3HT):PCBM mixture with certified light-to-energy efficiencies close to 4%.5 Since then, much effort has been directed towards the design and synthesis of novel low-band gap polymers with higher light absorption in the red (λabs > 600 nm). As a result of this intense research, a novel semiconductor polymer, namely PCPDTBT (poly1dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] was synthesized and used in OSC with outstanding light-to-energy conversion efficiency under 1 sun.6 Devices with efficiencies close to 5.5% have been recorded,7 and PCPDTBT has served as a platform for further improvements, leading to new polymers with higher performances in solar cell devices.3,8
It is of note to mention that in all the above-mentioned organic devices the electron acceptor moiety has always been a fullerene derivative such as C60-PCBM or C70-PCBM and, indeed, very little attention has been focused on replacing the fullerene derivative molecules by other acceptor moieties, either organic or inorganic in nature. As an exception, pioneering work on hybrid molecular solar cells utilizing semiconductor quantum dots as the electron acceptor material, has shown their potential in bulk-heterojunction solar cells with devices capable of efficiently converting sunlight into electrical power.9–11
Quantum dots (QD) have particular optical and electronic properties such as the possibility to fine tune their absorption and emission spectra by controlling the nanocrystal size.12 The stability of semiconductor quantum dots both under intense light and high temperatures, as well as their reversible electron transfer capabilities allowing them to donate or accept electrons and/or holes, make them ideal components in molecular solar cells.13,14 Furthermore, it has been postulated that semiconductor quantum dots can act as unique materials capable of carrying out, under light irradiation, multiple exciton generation processes, which would obviously boost solar cell efficiency.15
Remarkable advances on device efficiencies up to 2.7% at 1 sun in hybrid CdSe quantum dots/polymer solar cells have been recently published from different approaches.16–18 In fact, using branched CdSe semiconductor nanocrystals such as tetrapods, several research groups have measured efficiencies surpassing 3% at 1 sun.19 However, there is still a lack of fundamental understanding of the interfacial charge transfer processes that take place between the quantum dots and polymers,11 hence, the efficiency of such devices has not reached similar values as those mentioned above for (P3HT):PCBM based devices.
Several research groups, including our own, have carried out studies investigating the interfacial charge transfer reactions on polymer/QD films using photo-induced absorption spectroscopy (PIA)20–22 and laser transient absorption spectroscopy (L-TAS).23 In these recent studies, the electron recombination dynamics have been demonstrated to correspond to a multiple decay process due to the different energy traps present at the surface of the nanocrystal, and most of the measurements show pseudo-first order bimolecular recombination dynamics rather than the observed second-order recombination reaction in organic bulk-heterojunction solar cells. Yet, none of these studies have been carried out on complete devices but on thin films and therefore, it has proved difficult to relate such experimental results directly to photovoltaic device operation under continuous light irradiation.24,25
In this manuscript, we present a complete study in efficient PCPDTBT:CdSe quantum dot bulk heterojunction solar cells based on photo-induced steady-state carrier lifetimes and charge density measurements under continuously illuminated open-circuit conditions using the transient photovoltage (TPV) technique, complemented with the use of L-TAS to study the interfacial charge recombination in these devices. Our aim is to establish a rate law for the recombination processes, which limits the efficiency in CdSe quantum dot/polymer solar cells, as well as to shed more light on the aspects that will improve the device light-to-energy conversion efficiency.
Results and discussion
Device characterization
The CdSe QDs:PCPDTBT blend film UV-visible spectrum is shown in Fig. 1. As can be seen, the light absorption extends from the visible to the near IR region as expected from a low-band gap polymer. Moreover, in the UV-visible region the QD light absorption complements the polymer absorption. The spectrum shows a shoulder located at 610 nm, corresponding to the characteristic of the last excitonic peak of the CdSe QDs with an average diameter of 4.7 nm, and a second band with a maximum at 730 nm, which can be related to the absorption band of the PCPDTBT polymer, as has been reported previously.6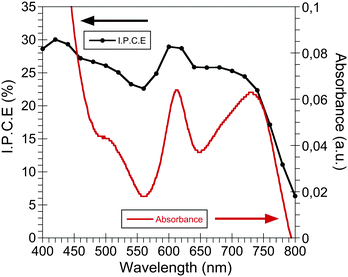 | ||
| Fig. 1 The UV-visible spectrum (red line) and the IPCE (black line) of a stable CdSe QDs:PCPDTBT bulk heterojunction device measured after 10 days after being manufactured. | ||
Furthermore, Fig. 1 also includes the incident photon to current conversion efficiency (IPCE) of one of our CdSe QDs:PCPDTBT solar cells. The IPCE shows a maximum photon to current conversion of 30%, but most important is the fact that clearly both device components, the CdSe QDs and the semiconductor polymer, are both capable of generating photocurrent.
The J–V characteristics at different light intensities are shown in Fig. 2a. The devices showed a maximum current density of 6.89 mA cm−2 and an open-circuit voltage 614 mV at 1 sun.
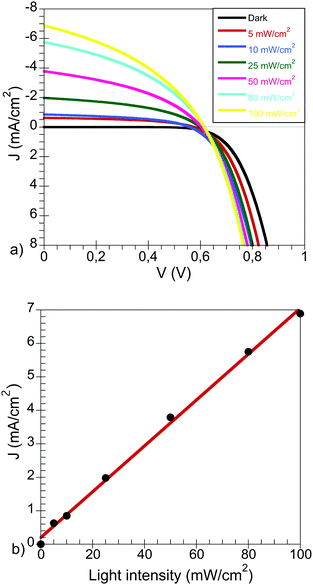 | ||
| Fig. 2 (a) J–V characteristics of a PCPDTBT:CdSe device at different light intensities and (b) dependence of JSC with the light intensity in these devices. | ||
The initial overall light-to-energy conversion efficiency was η = 1.9%, at 100 mW cm−2 sun-simulated light irradiation with measurements carried out for this work only made after the device physical parameters (Jsc, fill factor (FF) and Voc) remained constant.
In our opinion, this requisite is essential in order to be able to extract valuable general information in this type of devices. A summary of the J–V device characteristics is listed in Table 1.
| I/mW cm−2 | J sc/mA cm−2 | V oc/V | FF (%) | Efficiency (%) |
|---|---|---|---|---|
| 5 | 0.63 | 0.573 | 47.8 | 3.45 |
| 10 | 0.85 | 0.558 | 45.3 | 2.15 |
| 25 | 1.98 | 0.599 | 43.7 | 2.07 |
| 50 | 3.79 | 0.604 | 39.8 | 1.82 |
| 80 | 5.75 | 0.614 | 39.4 | 1.74 |
| 100 | 6.89 | 0.614 | 27.6 | 1.17 |
As can be seen, the photocurrent density is linear with the light intensity (Fig. 2b). This observation suggests that no major losses due to non-geminative recombination processes occur at short circuit. This behaviour has also been observed for organic solar cells in polymer bulk heterojunctions, where the dependence of the current density with the light intensity follows a power law function instead.26,27 In those cases a linear dependency indicates that the recombination is first order in carrier density (such as geminative recombination). In addition the open-circuit voltage on the devices is barely affected by the light intensity, as can be seen in Fig. 2a.
We will show that it is this accumulated free charge in the film that is responsible for setting the Fermi levels in the different materials and, therefore, the difference in Fermi level energies between the electron transport material and the hole transport material is responsible for the open-circuit voltage of the solar cell, as has been shown for other molecular solar cells and has been recently discussed in detail by Garcia-Belmonte and Bisquert.28 Hence, a direct correlation exists between charge accumulation, the electron lifetime and the device photovoltage in CdSe QDs:PCPDTBT solar cells.
Photo-induced charge recombination dynamics
Laser transient absorption spectroscopy (L-TAS) is used to study the photo-induced charge recombination dynamics in thin films such as in devices used for efficiency measurements. The UV-Vis region (inset) and the lifetime decay of radical cations (PCPDTBT polarons) in blend films29 with CdSe quantum dots are shown in Fig. 3. The decay of the transient signal in the microsecond–milisecond time scale can be fitted with a stretched exponential function (eqn (1)).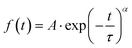 | (1) |
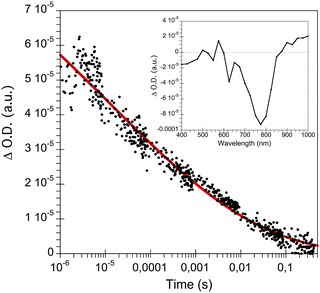 | ||
| Fig. 3 Transient absorption decay of CdSe QDs:PCPDTBT monitored at 980 nm after laser excitation at 535 nm. The inset corresponds to the transient absorption spectrum measured at 500 μs at the same excitation wavelength. | ||
From the fit we can extract the lifetime (τ) of the PCPDTBT polarons in the blend of 11 μs and the stretched exponential factor (α). This factor gives information about the dispersive of the system; α = 1 corresponds to truly monoexponential recombination kinetics, while with α < 1 there is more than one recombination pathway. In this case α = 0.13, suggesting that there are multiple decay processes in the blend film due to the different energies of the traps present in the bulk heterojunction. This result is in good agreement with the previous published results in polymer:CdSe quantum dots using photoinduced absorption spectroscopy (PIA).20,21 In general, the observation of stretched exponential decays in nanocrystal/polymer thin film devices from pseudo-first order recombination kinetics is in contrast to the power-law decays measured in bulk-heterojunction organic solar cells (see SI1, ESI†) that are assigned to second-order recombination processes.
Now, we turn to the charge carrier lifetime in complete devices under working conditions. The total charge in the device at different Voc corresponding to different light intensities (so called light bias) was measured by the charge extraction (CE) technique as described previously, and the results are shown in Fig. 4. We would like to stress the fact that the acquisition time used in the CE technique is the fastest procedure (minutes) to measure the device charge (Q) and the device capacitance (C), which implies that the device physical parameters (Jsc, Voc and FF) remain constant during the measurements, a unique property of this time-resolved technique.
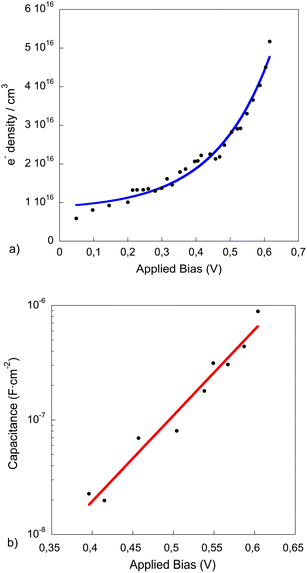 | ||
| Fig. 4 Electron density measured with the charge extraction technique (a) and capacitance (b) vs. light bias induced open-circuit voltage of complete device measured under standard operating conditions. The blue line in (a) and the red line in (b) correspond to the data fit to an exponential function. | ||
As illustrated in Fig. 4a, the experimental points can be fitted to an exponential equation. This exponential behaviour is usually observed when the photo-generated charges are being accumulated at the tail of the density of states (DOS) as the light intensity is raised. In contrast, a linear behaviour with the light bias (not observed for these devices at high light intensities) will imply that the charge is accumulated at the metal electrodes and the cell photovoltage is determined by the Vbi (built-in-field). The calculated geometrical capacitance for our devices was 15 nF cm−2 in the range determined experimentally for light bias below 0.4 V.
The data in Fig. 4b has been fitted to an exponential function (eqn (2)), with a value of the initial carrier concentration (n0 = 8.108 × 1015), when no light bias is applied, and γ = 9.2 between 0.4 and 0.62 V. These values have been obtained from the average of the data extracted from several devices and the good correlation of the experimental data to an exponential function implies that the device is highly p-doped as is the case in P3HT/PCBM organic solar cells (see SI2, ESI†), and the states in the tail of the DOS of the polymer are those which dominate the recombination rate.
n = n0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(γVoc) exp(γVoc) | (2) |
The chemical capacitance was also calculated from the charge extraction data. The differential capacitance C is defined as the voltage change ΔVoc when a small amount of charge ΔQ is added to the device (eqn (3)). These values are also shown in Fig. 4b.
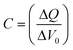 | (3) |
ΔQ may be obtained by integrating with respect to time the current measurement obtained for each light bias applied. Moreover, we have estimated the charges that may be lost before extraction due to their recombination during transport, and the result is that the amount of charge, which is not extracted, is negligible (less than 4%) and does not affect our results whatsoever. In any case, these losses will affect the magnitude of the curves: however the curve shape will be the same. Previous studies showed also an error lower than 10% when employing this technique on similar device structures.24,30
Fig. 4b shows the capacitance curve in the range from 400 to 620 mV. For lower values of voltage (Voc < 400 mV) there is no excess of charge accumulated in the device active layer and the capacitance obtained corresponds to the electrode capacitance, which has been found to be 15 nF cm−2 for our device, as mentioned before. The device built-in-voltage (VBi, flat-band conditions at the cathode) has been extracted from a Mott–Schottky plot (data not shown) and found to be 0.32 V. It should be noticed that the Vbi refers to a particular interface and it does not limit the device Voc. On the other hand, at light bias higher than 400 mV the capacitance is found to increase exponentially with Voc. Since the charge accumulation starts at potentials close to the Voc observed in the J–V curve at 1 sun, the light dependent charge recombination lifetimes were measured at these potentials.
The transient photovoltage (TPV) was measured over a range of light intensities corresponding to a Voc range of 0.4–0.62, see Fig. 5a, which is the same range we evaluate the accumulated charge. The lifetime of the carriers in a complete cell (τr) was found to vary exponentially with the Voc (eqn (4)).
| τr = τr0·exp(−βVoc) | (4) |
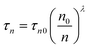 | (5) |
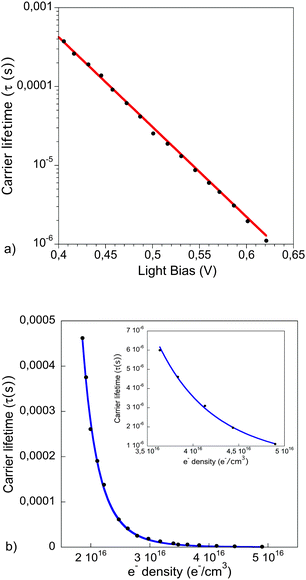 | ||
| Fig. 5 (a) Recombination lifetime at open circuit conditions extracted from fitting the transient photovoltage to a single exponential function, the red line corresponds to an exponential fit. (b) Carrier lifetime as a function of charge density with Voc from 400 to 620 mV. The blue line corresponds to the data fit to eqn (5). The inset shows a zoom of the fitted data points at high charge density. | ||
The charge carrier decay dynamics exhibit a strong carrier density dependency. The rate constant follows a power law function of charge density with an exponent λ = 3. This dependence is stronger than shown by polymer:fullerene or small molecule:fullerene solution processable solar cells (see SI3 in ESI†). In contrast, values between 3.7 and 4.3 have been shown in DSSC,31 depending on the electrolyte composition.
In the case of OPV, non-geminative recombination processes have been suggested to explain the inverse square relationship between the recombination rate and the charges, exhibiting a third-order dependence on charge density (dn/dt ∝ n3). On the other hand, a non-defined number of species in the electrolytes of the DSSC provide multiple recombination pathways, obtaining dispersive recombination dynamics.
In polymer:QD solar cells bimolecular recombination is also present in the device. The exponential dependence of the carrier lifetime with the applied bias, and the fact that the recombination under operating conditions follows a power law function, points this out. However, the stronger dependence of the carrier lifetime with the charge density indicates that, in addition to the bimolecular events that govern the recombination dynamics in OPV, there are other pathways in the bulk or between the photoactive materials and the electrodes that affect the recombination dynamics in these devices too.
In order to analyse the effect of electrode dependent processes such as non-selective contacts, shunt losses, or the presence of macroscopic electric fields in these devices we have compared, as shown in Fig. 6, the total charge carrier density (n) obtained with TPV/CE and the charge carrier density extracted from the L-TAS24,32 The L-TAS measurements were undertaken without the presence of device metal electrodes, hence the bulk heterojunction thin film is not affected by microscopic electric fields. Therefore, if both experimental sets of data: (1) the TPV/CE, which measurements are done in complete devices, and (2) L-TAS are in good agreement, we can conclude that the non-geminative charge recombination losses are not determined by any process dependent upon device electrodes at the time scale that our charge recombination measurements have been carried out (5 × 10−6–0.1 ms).
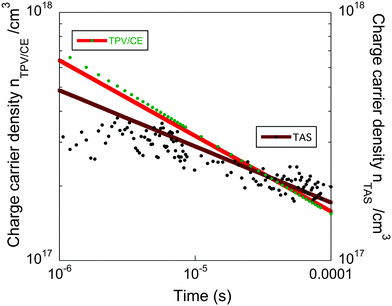 | ||
| Fig. 6 Comparison of charge carrier density (n) as function of time (t) derived from TPV/CE and L-TAS data. | ||
The experimental points were fitted to power law n(t) ∝ t−α where αTPV/CE ∼ 0.3 and αTAS ∼ 0.23. We observe similar results for both the TPV/CE data and the L-TAS measurements and thus we conclude that the non-geminative recombination dynamics in these devices are not influenced by the presence of processes dependent of the electrodes. We believe that the small variation observed in Fig. 6 can be attributed to the experimental error (see ESI†).
Therefore, the strong dependence of the recombination dynamics with the charge density on these devices in comparison with other OPV solar cells must be attributed to other factors (for example: nanomorphology) rather than electric fields due to the metal electrodes. However, further studies for recombination processes at earlier time scales, where the different mobility of the free carriers will play a major role, will be needed to shed more light on the influence of the metal contacts over the geminative charge recombination. The fact that the semiconductor nanocrystals are covered with capping ligands could also contribute to the generation of different recombination pathways, which would be different from fullerene:polymer OPV. Although usually capping ligands, such as phosphines derivatives or oleic acid, are believed to be removed from the quantum dot surface when interchange ligand reactions are carried out, the fact is that there is no evidence of their complete elimination. These organic molecules may play a role in the interfacial reactions between the nanocrystals and the polymers, and promote recombination processes that are not present in OPV.
Conclusions
In conclusion, we have demonstrated that in CdSe QDs:PCPDTBT bulk heterojunction solar cells the limiting factors in device working conditions can be studied using time-resolved spectroscopic techniques, and we found that the carrier recombination dynamics strongly depend upon the charge density and not on the electric fields which may be present close to the selective metal contacts. This dependence has been found to be higher than in organic solar cells, and indeed could be related to the inhomogeneous interface between the semiconductor nanocrystals and the polymer, due to the presence of capping agents and/or the defects at the surface of the nanocrystal. We would like to mention that previous work comparing CdSe/polymer and PCBM-C60/polymer cells have determined that the lifetime of the carriers are similar for both devices.33 However, such comparison was carried out in different conditions, as while the authors compared the device carrier lifetime at the same light intensities, they did not make a comparison at the same charge density. As we have demonstrated here, the carrier lifetime is very much dependent on the accumulated charge and, thus, in order to compare carrier lifetimes it is more useful to analyse the data at the same charge density which, indeed, could be different at the same light intensity.Acknowledgements
E. P. and J. A. would like to acknowledge the European Research Council starting Grant ERCstg-POLYDOT and the national projects CONSOLIDER HOPE 0007-2007 and MICINN CTQ-2007-60746-BQU as well as Catalonian regional government for the project 2009 SGR 207. The other authors gratefully acknowledge the financial support from the German federal Ministry of Education and research (BMBF) within the project “NanoPolySol” under contract No. 03X3517E. M. K. and M. E. additionally thank the German Research Foundation (DFG) graduate school GRK 1322 “Micro Energy Harvesting” for financial support.Notes and References
- Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032–6038 CrossRef CAS.
- P. Wang, S. M. Zakeeruddin, J. E. Moser, M. K. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402–407 CrossRef CAS.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS.
- R. F. Service, Science, 2011, 332, 293 CrossRef CAS.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef.
- I.-W. Hwang, S. Cho, J. Y. Kim, K. Lee, N. E. Coates, D. Moses and A. J. Heeger, J. Appl. Phys., 2008, 104, 033706 CrossRef.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS.
- D. S. Ginger and N. C. Greenham, Phys. Rev. B: Condens. Matter, 1999, 59, 10622–10629 CrossRef CAS.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425–2427 CrossRef CAS.
- E. Martinez-Ferrero, J. Albero and E. Palomares, J. Phys. Chem. Lett., 2010, 1, 3039–3045 CrossRef CAS.
- A. Kongkanad, K. Tvrdy, K. Takechi, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2008, 130, 4007–4015 CrossRef.
- M. Sykora, M. A. Petruska, J. Alstrum-Acevedo, I. Bezel, T. J. Meyer and V. I. Klimov, J. Am. Chem. Soc., 2006, 128, 9984–9985 CrossRef CAS.
- R. Debnath, J. Tang, D. A. Barkhouse, X. Wang, A. G. Pattantyus-Abraham, L. Brzozowski, L. Levina and E. H. Sargent, J. Am. Chem. Soc., 2010, 132, 5952–5953 CrossRef CAS.
- V. I. Klimov, J. Phys. Chem. B, 2006, 110, 16827–16845 CrossRef CAS.
- Y. Zhou, M. Eck, C. Veit, B. Zimmermann, F. Rauscher, P. Niyamakom, S. Yilmaz, I. Dumsch, S. Allard, U. Scherf and M. Krüger, Sol. Energy Mater. Sol. Cells, 2011, 95, 1232–1237 CrossRef CAS.
- W. Yu, H. Zang, Z. Fan, J. Zhang, H. Wei, D. Zhou, B. Xu, F. Li, W. Tian and B. Yang, Energy Environ. Sci., 2011, 4, 2831–2834 CAS.
- N. Radychev, I. Lokteva, F. Witt, J. Kolny-Olesiak, H. Borchert and J. Parisi, J. Phys. Chem. C, 2011, 115, 14111–14122 CAS.
- S. Dayal, N. Kopidakis, D. C. Olson, D. S. Ginley and G. Rumbles, Nano Lett., 2010, 10, 239–242 CrossRef CAS.
- P. Wang, A. Abrusci, H. M. P. Wong, M. Svensson, M. R. Andersson and N. C. Greenham, Nano Lett., 2006, 6, 1789–1793 CrossRef CAS.
- M. Pientka, J. Wisch, S. Boger, J. Parisi, V. Dyakonov, A. Rogach, D. Talapin and H. Weller, Thin Solid Films, 2004, 48, 451–452 Search PubMed.
- H. C. Leventis, S. P. King, A. Sudlow, M. S. Hill, K. C. Molloy and S. A. Haque, Nano Lett., 2010, 10, 1253–1258 CrossRef CAS.
- J. Albero, E. Martinez-Ferrero, J. Ajuria, C. Waldauf, R. Pacios and E. Palomares, Phys. Chem. Chem. Phys., 2009, 11, 9644–9647 RSC.
- C. G. Shuttle, B. O'Regan, A. M. Ballantyne, J. Nelson, D. D. C. Bradley, J.d. Mello and J. R. Durrant, Appl. Phys. Lett., 2008, 92, 093311 CrossRef.
- B. C. O'Regan and J. R. Durrant, J. Phys. Chem. B, 2006, 110, 8544–8547 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi, H. Xie and W. P. Blom, Appl. Phys. Lett., 2005, 87, 203502 CrossRef.
- G. Garcia-Belmonte, P. P. Boix, J. Bisquert, M. Sessolo and H. J. Bolink, Sol. Energy Mater. Sol. Cells, 2010, 94, 366–375 CrossRef CAS.
- J. Bisquert and G. Garcia-Belmonte, J. Phys. Chem. Lett., 2011, 2, 1950–1964 CrossRef CAS.
- I.-W. Hwang, C. Soci, D. Moses, Z. Zhu, D. Waller, R. Gaudiana, C. J. Brabec and A. J. Heeger, Adv. Mater., 2007, 19, 2307–2312 CrossRef CAS.
- A. Murano, R. Hamilton, C. G. Shuttle, A. M. Ballantyne, J. Nelson, B. O'Regan, W. Zhang, I. McCulloch, H. Azimi, M. Morana, C. J. Brabec and J. R. Durrant, Adv. Mater., 2010, 22, 4987–4992 CrossRef.
- B. C. O'Regan, J. R. Durrant, P. M. Sommeling and N. J. Bakker, J. Phys. Chem. C, 2007, 111, 14001–14019 CAS.
- A. Maurano, C. G. Shuttle, R. Hamilton, A. M. Ballantyne, J. Nelson, W. Zhang, M. Heeney and J. R. Durrant, J. Phys. Chem. C, 2011, 115, 5947–5957 CAS.
- Z. Li, F. Gao, N. C. Greenham and C. R. McNeill, Adv. Funct. Mater., 2011, 21, 1419–1431 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectroscopic characterization and L-TAS and CE/TPV comparison. See DOI: 10.1039/c1sc00514f |
| This journal is © The Royal Society of Chemistry 2011 |