Pd nanocrystals with single-, double-, and triple-cavities: facile synthesis and tunable plasmonic properties†
Zhiqiang
Niu
a,
Yu-Rong
Zhen
b,
Ming
Gong
a,
Qing
Peng
a,
Peter
Nordlander
b and
Yadong
Li
*a
aDepartment of Chemistry, Tsinghua University, Beijing, 100084, P.R. China. E-mail: ydli@mail.tsinghua.edu.cn; Fax: +86-10-62788765; Tel: +86-10-62772350
bDepartment of Physics and Astronomy, M.S. 61, Rice University, Houston, Texas 77005-1892, USA
First published on 22nd September 2011
Abstract
The surface plasmon resonance (SPR) of palladium (Pd) nanocrystals is of potential importance due to their catalytic properties and surface stability. Pd nanocrystals with single-, double-, and triple-cavities were produced using a one-pot aqueous approach, where micelles formed by PVP dictate the cavity formation. The cavity number and cavity size were further regulated by systematically changing the experimental parameters. The SPR of the as-prepared hollow Pd can be tuned from 355 nm to 702 nm to cover the entire visible spectral region. The plasmonic nature of the hollow Pd was investigated by finite-difference-time-domain simulation, which indicated that the SPR shift was mainly attributed to the size variations of the monomer cavity.
Introduction
The SPR of metallic nanoparticles is the foundation for many applications, such as spectral signal enhancement, photothermal transduction, biomedical imaging, and plasmonic waveguiding.1 A crucial requisite for optimizing such applications is achieving spectral control of the SPR wavelengths.1g The SPR bands of Au and Ag nanoparticles of different shapes have been studied intensively and can be systematically tuned from the visible to the near-infrared.1c,2 The SPR features of Pd are potentially also important.3 For example, Pd nanosheets have been shown to be a promising candidate for cancer therapy due to their photothermal stability.3a Moreover, it is possible to observe a catalytic process in situ by combining the catalytic and plasmonic properties of Pd nanoparticles.3b However, the SPR bands of pure Pd nanoparticles are usually located in the near-UV region,4 and the fabrication of Pd nanoparticle geometries with tunable SPR in the whole visible region has received limited success.5Metallic shell structures represent an important class of SPR tunable nanoparticles because their plasmon resonances depend strongly on the ratio of the shell thickness to the overall diameter.6 Hollow metallic nanoshells can be prepared by hard templating methods,7 where a metallic shell is grown around a presynthesized core (e.g., silica particles, carbon spheres, polymer latex colloids) which is removed after the growth of the shell. While the size distributions of such nanoshell particles can be well controlled, the multi-step nature of this fabrication procedure is somewhat complicated. The galvanic replacement approach is another method to prepare hollow metallic nanocrystals.4b,8 Here reducible metal ions are deposited on the surface of a foreign metal template (e.g., Ag or Co nanoparticles), followed by the oxidation and dissolution of the template to generate hollow cavities. Although the sacrificial metal templates need no additional removal procedure, they may introduce impurities to the system in the form of alloys or adatoms.8d,9 In the past decade, micelle-directed synthesis of hollow nanostructures has attracted enormous interest due to the advantages of easy removal of templates and facile encapsulation of functional species. Lots of hollow oxides have been successfully made in this way.10 However, extension of this method to metallic shell structures still lacks exploration.11
Furthermore, complex metallic shell structures constructed from the assembly of elementary geometries are of great significance due to their distinct plasmon resonance behaviors. Taking pyramidal Au nanoshells for instance, it has been shown that both the stacked and side-to-side nanopyramid dimers/trimers produce stronger surface enhanced Raman scattering (SERS) responses than monomers.12 Besides, the resonant frequencies of multishelled Au nanospheres have been demonstrated to be determined by the hybridization of the plasmons of the inner and outer shells.6 To the best of our knowledge, fabrication of metallic shell structures with multiple cavities still remains a challenging task and their plasmonic properties need further investigation.
In this edge article, we present a facile, one-pot approach for the fabrication of hollow Pd nanostructures with single-, double-, and triple-cavities. The growth mechanism of these hollow structures was investigated by time-dependent transmission electron microscopy (TEM) studies. The SPR properties of the as-obtained hollow Pd nanocrystals were subsequently studied by UV–vis spectroscopy and theoretical simulations.
Results and discussion
In a typical synthesis of Pd nanoshells, Pd(NH3)4Cl2 was used as metal precursor, PVP (polyvinyl pyrrolidone) was employed as surfactant, reductant, and cavity-dictator concurrently, and water was selected as solvent. The reaction was carried out by mixing these chemicals in a 10 mL autoclave and heating them at 200 °C for 2 h. The most striking feature of this synthetic system is its simplicity.Fig. 1a shows a large-area TEM image of such Pd nanocrystals. Generally, they have an average size of 56 nm (standard deviation: 7.5).13 Upon increased magnification (Fig. 1b), cavity structures can clearly be observed in the centers of the nanocrystals. These void cavities can have different forms, and can be qualitatively described as monomer, dimer, and trimer configurations of elementary spherical voids. Fig. 1c gives the cavity distributions based on a sample size of 623, where 56%, 34%, and 8% of the hollow Pd nanocrystals are with single-, double-, and triple-cavities, respectively. A high-resolution TEM image of a nanoshell (Fig. S1a†) shows clear lattice fringes with a spacing of 0.23 nm, corresponding to the {111} interplanar distance of face centered cubic (fcc) Pd. The energy dispersive X-ray spectroscopy (EDS) line-scanning profile across a single Pd nanocrystal confirms its hollow feature (Fig. S1b†). Fig. 1d shows the corresponding powder XRD patterns, which confirm the fcc structure of Pd (JCPDS 65-6174).
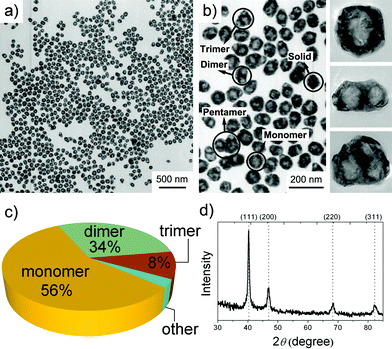 | ||
| Fig. 1 (a, b) TEM images, (c) cavity distribution pie chart, (d) powder XRD pattern of hollow Pd nanostructures. | ||
To understand the formation mechanism of the hollow Pd nanocrystals, a time-dependent experiment was performed by collecting product every 5 min. During the first 40 min, the reaction solution remained colorless, indicating that the reaction had not started. However, some micelle-like nanostructures were observed as shown in Fig. S2.† In a 45 min reaction, the solution turned light brown, indicating the start of reduction. Small Pd nanoparticles with diameters of 5–20 nm were produced (Fig. 2a) by this time point. These small nanoparticles aggregate together and exhibit an incomplete hollow spherical morphology. In the following 5 min, these semi-mature hollow spheres continued to grow and developed intact profiles as depicted in Fig. 2b. The average external diameter of the hollow aggregates is about 227 nm, while the inner diameter is 136 nm. In a 60 min reaction (Fig. 2c), the dimensions of the hollow aggregates are significantly reduced to 129 nm (outer diameter) with a 48 nm inner diameter, which means the aggregates are more compact compared to the loose structures in Fig. 2b. Moreover, we found that some of the aggregates coupled with each other to form dimers as marked by arrows in Fig. 2c. These coupled structures will eventually result in dimer-cavity nanoparticles. Fig. 2d shows the TEM image after a 70 min reaction, where crystalline hollow nanospheres are produced. The observed transformation from small nanoparticle aggregates to larger crystalline structures can likely be attributed to an imperfect oriented attachment process, which has been used to account for the morphological evolution of nanocrystalline aggregates under hydrothermal conditions. Adjacent small nanoparticles attach together at the interface by sharing a common crystallographic orientation to form larger crystalline structures.14
 | ||
| Fig. 2 TEM images of products produced in (a) 45, (b) 50, (c) 60, and (d) 70 min reactions, respectively. (e) Proposed growth mechanism of nanoshells. | ||
PVP micelle-directed synthesis of hollow nanostructures has been reported in vanadium oxide and lead oxide.15 Based on these pioneering works and the above observations, we propose a probable three-step mechanism for the formation of Pd hollow structures, which is simplified as “reduction–aggregation–attachment”. As shown in Fig. 2e, PVP forms micelles in water. Pd ions are reduced by hydroxyl end groups of PVP to small nanoparticles around PVP micelles.16 As the reaction proceeds, more and more small nanoparticles are produced and form hollow aggregates. These aggregates further fuse into crystalline hollow nanospheres via imperfect oriented structures. It is worth noting that the hollow aggregates exhibit a random walk in the solution, and the collision of two or more aggregates leads to the formation of coupled structures as observed in Fig. 2c, which plays a key role in determining the multiple-cavity geometry of the final products (Scheme. S1†).
In principle, the stability of micelles depends on many parameters, such as temperature, pH, ionic strength, concentration of surfactant, etc., restricting the micelle-based synthesis to certain experimental conditions.9a,17 To test the flexibility of our synthetic protocol, we first changed the quantity of PVP from 0.1 g to 0.025 g and 0.4 g respectively (Fig. S3†). The size of the nanoparticle products decreased slightly with the increase of PVP quantity (Fig. 3a), specifically, 58 nm for 0.025 g PVP, 56 nm for 0.1 g PVP, 52 nm for 0.4 g PVP. The fraction of multiple-cavity Pd nanoparticles increased dramatically with increasing concentration of PVP (Fig. 3b). The percentages of monomer, dimer, and trimer were 85%, 11%, and 3% in the 0.025 g PVP system, while in the 0.4 g PVP system they were 45%, 39%, and 13%, respectively. As mentioned above, the multiple-cavity Pd nanostructures result from collisions of hollow aggregates. An increase in PVP concentration will thus enhance the probability of such collisions, resulting in a higher yield of multiple-cavity Pd nanoparticles. We then attempted to separate the dimers and trimers by density gradient centrifugation using either CsCl aqueous solution or water–iodixanol solution as the gradient medium but failed in both cases. Fig. 3c shows the UV–vis spectra of Pd nanocrystals produced with 0.025, 0.1, and 0.4 g PVP. The SPR absorption peaks blue-shift with increase of PVP concentration.
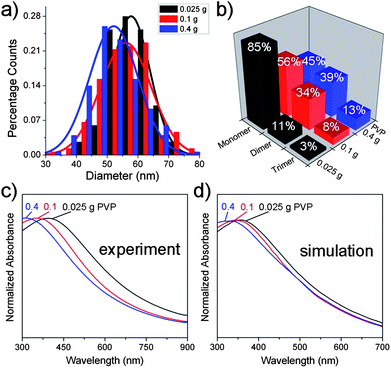 | ||
| Fig. 3 Particle size distribution histograms (a) and cavity distributions (b) of hollow Pd nanoparticles produced with 0.025 g, 0.1 g, and 0.4 g PVP. Note that the diameters slightly decreased from 58 nm to 52 nm while multiple-cavity Pd nanoparticles increased dramatically. Experimental (c) and calculated (d) UV–vis spectra of hollow Pd nanocrystals synthesized using 8 mM metal precursors and 0.025 g, 0.1 g, and 0.4 g PVP. | ||
The plasmonic nature of the Pd nanoshells can be modeled using numerical calculations, Mie for monomers and finite-difference-time-domain (FDTD) for dimers, described in the ESI,† with the optical constants for palladium obtained from the literature.18 The calculated normalized absorption spectra for Pd nanoparticles for 0.025, 0.1, and 0.4g PVP, presented in Fig. 3d, reproduce the experimentally observed blue shift with increasing PVP concentration. The peak positions of the spectra from mixed monomer–dimer aggregates are found to be very close to those from only monomer cavities, indicating that the blue shift of the SPR is mainly attributed to the size variation of the monomer cavity with PVP concentration. With diminishing core radius and almost unchanged shell thickness (Table S1†), the SPR of a nanoshell exhibits a blue-shift due to plasmon hybridization.6
We also found that the particle size could be regulated by varying the concentration of metal precursor from 2 to 24 mM, specifically, 2 mM for (16 ± 3) nm, 4 mM for (29 ± 4) nm, 8 mM for (56 ± 8) nm, 12 mM for (61 ± 7) nm, 16 mM for (76 ± 10) nm, 20 mM for (88 ± 10) nm, and 24 mM for (168 ± 13) nm (Fig. S4†). The standard deviation of particle sizes gradually increased due to the declining molar ratio of PVP repeating units to metal precursors (Fig. 4a). Fig. 4b depicts the UV–vis spectra of Pd nanocrystals prepared from 2 to 24 mM metal precursors. The spectra show a clear red-shift from 355 nm to 702 nm covering most of the visible region of the spectrum. To the best of our knowledge, this is the first time that Pd nanostructures have been synthesized by wet chemical method that display tunable SPR properties over such a wide spectral range.
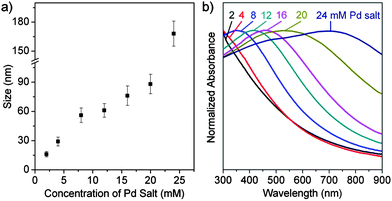 | ||
| Fig. 4 (a) The size of hollow Pd nanocrystals as a function of the concentration of Pd salt. (b) UV–vis spectra of hollow Pd nanocrystals synthesized by using 0.1 g PVP and 2, 4, 8, 12, 16, 20, and 24 mM metal precursors. | ||
Conclusions
In summary, a facile micelle-directed protocol was employed to fabricate hollow Pd nanocrystals with single-, double-, and triple-cavity structures. The cavity and size distributions of the hollow Pd nanocrystals can be tuned by changing the concentrations of surfactant and metal precursor, respectively. The resulting Pd nanoshells display a SPR which is tunable across the whole visible region, providing a novel platform for applications in probing catalytic reactions and photothermal therapy. The plasmonic nature of these hollow particles with great geometrical complexity was also studied via several calculations. The reported synthesis is simple and flexible, and may be extended to other metallic shell structures.Acknowledgements
This work was supported by the State Key Project of Fundamental Research for Nanoscience and Nanotechnology (2011CB932401), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (Grant No. 20921001), and the Robert A Welch foundation (C-1222).Notes and references
- (a) Z. Q. Tian, B. Ren and D. Y. Wu, J. Phys. Chem. B, 2002, 106, 9463 CrossRef CAS; (b) S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842 CrossRef CAS; (c) P. K. Jain, X. H. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578 CrossRef CAS; (d) J. Chen, F. Saeki, B. J. Wiley, H. Cang, M. J. Cobb, Z. Y. Li, L. Au, H. Zhang, M. B. Kimmey, X. D. Li and Y. N. Xia, Nano Lett., 2005, 5, 473 CrossRef CAS; (e) A. F. Koenderink, Nano Lett., 2009, 9, 4228 CrossRef CAS; (f) T. Som and B. Karmakar, Nano Res., 2009, 2, 607 CrossRef CAS; (g) N. C. Bigall, T. Hartling, M. Klose, P. Simon, L. M. Eng and A. Eychmuller, Nano Lett., 2008, 8, 4588 CrossRef CAS; (h) A. M. Schwartzberg and J. Z. Zhang, J. Phys. Chem. C, 2008, 112, 10323 CrossRef CAS.
- (a) S. E. Skrabalak, J. Y. Chen, Y. G. Sun, X. M. Lu, L. Au, C. M. Cobley and Y. N. Xia, Acc. Chem. Res., 2008, 41, 1587 CrossRef CAS; (b) L. Au, Y. C. Chen, F. Zhou, P. H. C. Camargo, B. Lim, Z. Y. Li, D. S. Ginger and Y. N. Xia, Nano Res., 2008, 1, 441 CrossRef CAS; (c) A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935 CrossRef CAS.
- (a) X. Huang, S. Tang, X. Mu, Y. Dai, G. Chen, Z. Zhou, F. Ruan, Z. Yang and N. Zheng, Nat. Nanotechnol., 2010, 6, 28 CrossRef; (b) K. N. Heck, B. G. Janesko, G. E. Scuseria, N. J. Halas and M. S. Wong, J. Am. Chem. Soc., 2008, 130, 16592 CrossRef CAS.
- (a) Y. J. Xiong, J. Y. Chen, B. Wiley, Y. A. Xia, Y. D. Yin and Z. Y. Li, Nano Lett., 2005, 5, 1237 CrossRef CAS; (b) J. Y. Chen, B. Wiley, J. McLellan, Y. J. Xiong, Z. Y. Li and Y. N. Xia, Nano Lett., 2005, 5, 2058 CrossRef CAS.
- C. Langhammer, B. Kasemo and I. Zoric, J. Chem. Phys., 2007, 126, 194702 CrossRef.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419 CrossRef CAS.
- (a) S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS; (b) C. Graf and A. van Blaaderen, Langmuir, 2002, 18, 524 CrossRef CAS.
- (a) S. J. Guo, S. J. Dong and E. Wang, Chem.–Eur. J., 2008, 14, 4689 CrossRef CAS; (b) Y. D. Yin, C. Erdonmez, S. Aloni and A. P. Alivisatos, J. Am. Chem. Soc., 2006, 128, 12671 CrossRef CAS; (c) H. P. Liang, L. J. Wan, C. L. Bai and L. Jiang, J. Phys. Chem. B, 2005, 109, 7795 CrossRef CAS; (d) H. P. Liang, Y. G. Guo, H. M. Zhang, J. S. Hu, L. J. Wan and C. L. Bai, Chem. Commun., 2004, 1496 RSC; (e) H. P. Liang, H. M. Zhang, J. S. Hu, Y. G. Guo, L. J. Wan and C. L. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540 CrossRef CAS; (f) S. Preciado-Flores, D. C. Wang, D. A. Wheeler, R. Newhouse, J. K. Hensel, A. Schwartzberg, L. H. Wang, J. J. Zhu, M. Barboza-Flores and J. Z. Zhang, J. Mater. Chem., 2011, 21, 2344 RSC.
- (a) Y. Vasquez, A. K. Sra and R. E. Schaak, J. Am. Chem. Soc., 2005, 127, 12504 CrossRef CAS; (b) X. Q. Huang, H. H. Zhang, C. Y. Guo, Z. Y. Zhou and N. F. Zheng, Angew. Chem., Int. Ed., 2009, 48, 4808 CrossRef CAS.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS.
- (a) D. B. Zhang, L. M. Qi, J. M. Ma and H. M. Cheng, Adv. Mater., 2002, 14, 1499 CrossRef CAS; (b) V. S. Murthy, J. N. Cha, G. D. Stucky and M. S. Wong, J. Am. Chem. Soc., 2004, 126, 5292 CrossRef CAS.
- K. A. Stoerzinger, W. Hasan, J. Y. Lin, A. Robles and T. W. Odom, J. Phys. Chem. Lett., 2010, 1, 1046 CrossRef CAS.
- The cross-section diameters of dimers and trimers were used to give a rough estimate of the product size distribution.
- (a) R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549 CrossRef CAS; (b) R. L. Penn and J. F. Banþeld, Science, 1998, 281, 969 CrossRef CAS.
- (a) A. M. Cao, J. S. Hu, H. P. Liang and L. J. Wan, Angew. Chem., Int. Ed., 2005, 44, 4391 CrossRef CAS; (b) G. C. Xi, Y. Y. Peng, L. Q. Xu, M. Zhang, W. C. Yu and Y. T. Qian, Inorg. Chem. Commun., 2004, 7, 607 CrossRef CAS.
- Y. Xiong, I. Washio, J. Chen, H. Cai, Z. Y. Li and Y. Xia, Langmuir, 2006, 22, 8563 CrossRef CAS.
- (a) H. L. Xu and W. Z. Wang, Angew. Chem., Int. Ed., 2007, 46, 1489 CrossRef CAS; (b) C. C. Huang, J. R. Hwu, W. C. Su, D. B. Shieh, Y. Tzeng and C. S. Yeh, Chem.–Eur. J., 2006, 12, 3805 CrossRef CAS; (c) F. Q. Guo, Z. F. Zhang, H. F. Li, S. L. Meng and D. Q. Li, Chem. Commun., 2010, 46, 8237 RSC; (d) Y. Liu, Y. Chu, Y. J. Zhuo, L. H. Dong, L. L. Li and M. Y. Li, Adv. Funct. Mater., 2007, 17, 933 CrossRef CAS; (e) X. J. Zhang and D. Li, Angew. Chem., Int. Ed., 2006, 45, 5971 CrossRef CAS.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1974, 9, 5056 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, FDTD simulation, and further TEM characterization. See DOI: 10.1039/c1sc00449b |
| This journal is © The Royal Society of Chemistry 2011 |