The first example of the direct asymmetric conjugate addition of aldehydes to a methylenemalonate promoted by an axially chiral amino diol catalyst†
Taichi
Kano
a,
Fumitaka
Shirozu
a,
Koichi
Tatsumi
a,
Yasushi
Kubota
b and
Keiji
Maruoka
*a
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto 606-8502, Japan. E-mail: maruoka@kuchem.kyoto-u.ac.jp; Fax: +81-75-753-4041; Tel: +81-75-753-4041
bNippon Soda Co., Ltd, 345, Takada, Odawara, Kanagawa 250-0280, Japan
First published on 26th August 2011
Abstract
A methylenemalonate could be employed as a reactive equivalent of a three carbon Michael acceptor such as acrylate in a direct asymmetric conjugate addition of aldehydes catalyzed by an axially chiral amino diol. The obtained conjugate addition product was readily converted to synthetically useful and important chiral building blocks.
Organocatalysis is a highly promising tool for the rapid construction of chiral building blocks in the asymmetric synthesis of natural products and biologically active compounds,1 and a wide variety of asymmetric conjugate additions through enamine intermediates have been developed.2,3 To the best of our knowledge, however, simple and versatile three-carbon Michael acceptors such as acrylate, acrylamide and acrylonitrile have not been employed in enamine catalysis, probably due to their low reactivity.4,5 In this context, methylenemalonates would be expected to be a reactive equivalent of acrylates,6,7 and conjugate addition products of this Michael acceptor as a three carbon unit would be of high value for multiple synthetic applications. However, methylenemalonates are known to react readily with amines,8 and enamine formation between an amine organocatalyst and an aldehyde would be inhibited by the competitive conjugate addition of the amine organocatalyst to methylenemalonates (Scheme 1). Previous studies in our laboratory demonstrated that binaphthyl-based secondary amines of type (S)-3 were less nucleophilic compared to typical pyrrolidine catalysts such as proline. Accordingly, we have been interested in the development of a direct asymmetric conjugate addition of aldehydes to methylenemalonate using this type of less nucleophilic secondary amine catalyst.
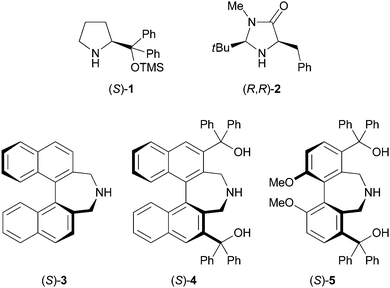
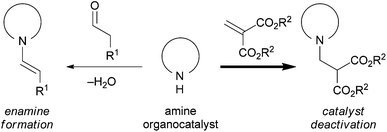
We first investigated conjugate addition of 3-phenylpropanal to di-tert-butyl methylenemalonate 6 in THF in the presence of various secondary amine catalysts (10 mol%), and the results are shown in Table 1. The use of pyrrolidine as a catalyst did not afford the desired conjugate addition product 7 (entry 1). Among representative chiral amine catalysts such as L-proline, (S)-19 and (R,R)-2,10 the reaction with (S)-1 gave the conjugate adduct 7 enantioselectively albeit in moderate yield (entry 3).11 When the less nucleophilic binaphthyl-based secondary amine (S)-3 was employed as a catalyst, the conjugate adduct 7 was not obtained (entry 5). In the NMR analysis, the conjugate addition of (S)-3 to 6 was observed. With the expectation of minimizing undesired catalyst deactivation by changing the steric environment of (S)-3, we considered using catalyst (S)-4 which has bulky hydroxydiphenylmethyl groups at 3,3′-positions.12 With (S)-4 having acidic hydroxy groups, the activation of 6 through hydrogen bonding would also be expected. Fortunately, the reaction of 3-phenylpropanal with 6 was found to be catalyzed by (S)-4 to give 7 in excellent yield with moderate enantioselectivity (entry 6). Use of a newly designed biphenyl-based catalyst (S)-5 resulted in higher enantioselectivity (entry 7). Encouraged by this promising result, the reaction conditions were optimized. Further improvement of enantioselectivity was achieved by lowering the reaction temperature to 0 °C (entry 8). With (S)-5, reactions were conducted in various solvents, and Et2O was found to be the optimal solvent in terms of both yield and enantioselectivity (entries 8–14). Even at low catalyst loading (3 mol%), the reaction proceeded smoothly to give 7 in good yield without affecting enantioselectivity (entry 16), while the reaction with 3 mol% of (S)-1 resulted in a significant decrease in yield (entry 17).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T/°C | Yield (%)b | ee (%)c |
| a The reaction of 3-phenylpropanal (0.4 mmol) with 6 (0.13 mmol) in a solvent was carried out in the presence of a catalyst (0.013 mmol) for 4 h. b The conjugate addition product was isolated as the corresponding alcohol to determine the enantioselectivity. c The ee of the product was determined by HPLC analysis using a chiral column. d Use of 3 mol% of a catalyst. | |||||
| 1 | Pyrrolidine | THF | rt | 0 | — |
| 2 | L-Proline | THF | rt | 48 | −6 |
| 3 | (S)-1 | THF | rt | 69 | −89 |
| 4 | (R,R)-2 | THF | rt | 0 | — |
| 5 | (S)-3 | THF | rt | 0 | — |
| 6 | (S)-4 | THF | rt | 99 | 68 |
| 7 | (S)-5 | THF | rt | 93 | 76 |
| 8 | (S)-5 | THF | 0 | 74 | 87 |
| 9 | (S)-5 | CH2Cl2 | 0 | 0 | — |
| 10 | (S)-5 | Toluene | 0 | 0 | — |
| 11 | (S)-5 | Dioxane | 0 | 61 | 87 |
| 12 | (S)-5 | CPME | 0 | 83 | 94 |
| 13 | (S)-5 | TBME | 0 | 68 | 93 |
| 14 | (S)-5 | Et2O | 0 | 94 | 94 |
| 15 | (S)-5 | Et2O | −20 | 71 | 95 |
| 16d | (S)-5 | Et2O | 0 | 83 | 95 |
| 17d | (S)-1 | Et2O | 0 | 38 | −97 |
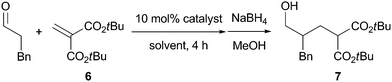
In the presence of 10 mol% of (S)-5, the direct asymmetric conjugate addition to 6 of several other aldehydes was examined, and the results are shown in Table 2. In general, these direct asymmetric conjugate additions gave the corresponding adducts with good to excellent enantioselectivity. The reactions with a lower catalyst loading (3 mol%) also gave satisfactory results (entries 2 and 6).
|
|
||||
|---|---|---|---|---|
| Entry | R | Conditions (°C, h) | Yield (%)b | ee (%)c |
| a Unless otherwise specified, the reaction of an aldehyde (0.4 mmol) with 6 (0.13 mmol) in Et2O was carried out for 4 h at 0 °C in the presence of (S)-5 (0.013 mmol). b Isolated yield. c The ee of the product was determined by HPLC analysis using a chiral column. d Use of 3 mol% of (S)-5. e Use of 1.5 equiv. of aldehyde. f The reaction was performed in THF. | ||||
| 1 | Me | 0, 4 | 80 | 96 |
| 2d | Me | 0, 4 | 79 | 96 |
| 3e | Bu | 0, 4 | 83 | 94 |
| 4e | Hex | 0, 4 | 83 | 94 |
| 5 | Bn | 0, 4 | 94 | 94 |
| 6d | Bn | 0, 4 | 83 | 95 |
| 7e | CH2Cy | 0, 4 | 70 | 86 |
| 8 | CH2OBn | 0, 4 | 69 | 93 |
| 9 | (CH2)3OBn | 0, 4 | 94 | 95 |
| 10e,f | CH2CO2Me | 0, 40 | 65 | 87 |
| 11e,f | CH2NHZ | 0, 40 | 82 | 84 |
| 12e | 3-Pyridylmethyl | 0, 4 | 87 | 93 |
| 13 | i-Pr | rt, 24 | 80 | 97 |
| 14e | Cy | rt, 72 | 89 | 94 |

The obtained conjugate adduct 8 was a versatile intermediate in organic synthesis and readily converted to important chiral building blocks (Scheme 2). When one-pot conjugate addition–reduction product 7 was treated with TFA at 80 °C, δ-lactone 9 was obtained in quantitative yield with complete retention of stereochemistry. Treatment of 7 with trifluoromethanesulfonyl chloride and DBU formed cyclic ether 10 without loss of optical purity.13 The conjugate addition product 8 could also be converted in situ to amine 11 by reductive amination. Amine 11 was transformed to δ-lactam 12 in good yield using TFA. In the presence of (diacetoxyiodo)benzene and tetrabutylammonium iodide, amine 11 was directly cyclized to pyrrolidine 13.14 Moreover, cyclohexene 14 was obtained in one-pot from the reaction of 8 with NaH and triphenyl(vinyl)phosphonium bromide.15
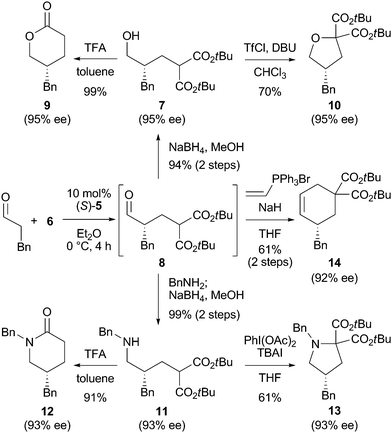 | ||
| Scheme 2 Transformations of the conjugate adduct 8. | ||
The synthetic utility of the present conjugate addition was successfully demonstrated in the formal synthesis of a glycinamide ribonucleotide formyltransferase inhibitor, pelitrexol,16,17 which has a potent antiangiogenesis activity, as shown in Scheme 3. Aldehyde 15 was converted in one-pot to amine 16 by conjugate addition and following reductive amination. Transesterification of 16 with H2SO4 in ethanol gave the corresponding ethyl ester 17 without affecting the benzyl ester. Treatment of 17 with Pd/C under a hydrogen atmosphere in acetic acid gave the deprotected δ-lactam 18 with an exthoxycarbonyl group attached to its α-position. Since 18 was an intermediate in a previous total synthesis of pelitrexol,17 this work contributes to its formal synthesis.
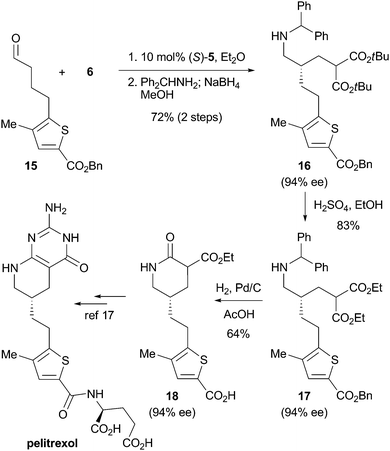 | ||
| Scheme 3 Formal total synthesis of pelitrexol. | ||
The absolute configuration of the product obtained in the present conjugate addition was determined by comparison of its optical rotation with the value in the literature.18 Based on the observed stereochemistry, a plausible transition state model can be proposed as shown in Fig. 1. Activated and directed by a hydroxyl group on (S)-5, 6 approaches the Re face of the enamine.
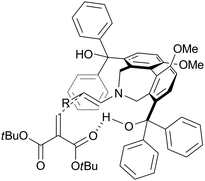 | ||
| Fig. 1 A transition state model for the direct asymmetric conjugate addition catalyzed by (S)-5. | ||
In summary, we have developed a direct asymmetric conjugate addition of aldehydes to methylenemalonate catalyzed by the novel axially chiral amino diol (S)-5. The conjugate addition product obtained is a versatile intermediate and could be readily converted to synthetically useful and important chiral building blocks. We are currently applying this methodology toward the synthesis of biologically active compounds and pharmaceutical targets.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from MEXT, Japan. F.S. thanks the Japan Society for the Promotion of Science for Young Scientists for Research Fellowships.Notes and references
- For reviews, see: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (c) A. Berkessel and H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (d) Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed.
- For reviews, see: (a) M. Marigo and K. A. Jørgensen, Chem. Commun., 2006, 2001 RSC; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS.
- For recent reviews on organocatalytic asymmetric conjugate additions, see: (a) G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79 CAS; (b) J. L. Vicario, D. Badía and L. Carrillo, Synthesis, 2007, 2065 CrossRef CAS; (c) D. Almasi, D. A. Alonso and C. Najera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS; (d) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS; (e) S. Sulzer-Mossé and A. Alexakis, Chem. Commun., 2007, 3123 RSC.
- The reaction between an enamine and methyl acrylate requires a high temperature and long reaction time. See: G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz and R. Terrell, J. Am. Chem. Soc., 1963, 85, 207 CrossRef CAS.
- For highly enantioselective organocatalytic conjugate addition of aldehydes to simple enones with four or more carbons in the chain, see: (a) T. J. Peelen, Y. Chi and S. H. Gellman, J. Am. Chem. Soc., 2005, 127, 11598 CrossRef CAS; (b) Y. Chi and S. H. Gellman, Org. Lett., 2005, 7, 4253 CrossRef CAS.
- For organocatalytic asymmetric conjugate additions to α-substituted acrylates and β-substituted methylenemalonates, see: (a) O. Penon, A. Carlone, A. Mazzanti, M. Locatelli, L. Sambri, G. Bartoli and P. Melchiorre, Chem.–Eur. J., 2008, 14, 4788 CrossRef CAS; (b) Ł. Albrecht, B. Richter, H. Krawczyk and K. A. Jørgensen, J. Org. Chem., 2008, 73, 8337 CrossRef; (c) G.-L. Zhao, J. Vesely, J. Sun, K. E. Christensen, C. Bonneau and A. Córdova, Adv. Synth. Catal., 2008, 350, 657 CrossRef CAS; (d) L. Wen, Q. Shen and L. Lu, Org. Lett., 2010, 12, 4655 CrossRef CAS.
- M. E. Kuehne and P. J. Reider, J. Org. Chem., 1985, 50, 1464 CrossRef CAS.
- (a) R. E. Gawley, S. Pinet, C. M. Cardona, P. K. Datta, T. Ren, W. C. Guida, J. Nydick and R. M. Leblanc, J. Am. Chem. Soc., 2002, 124, 13448 CrossRef CAS; (b) S. Guillarme, S. Legoupy, A.-M. Aubertin, C. Olicard, N. Bourgougnon and F. Huet, Tetrahedron, 2003, 59, 2177 CrossRef CAS.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS; (b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS.
- S. P. Brown, N. C. Goodwin and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192 CrossRef CAS.
- The NMR analysis revealed that pyrrolidine in THF-d8 was completely consumed by equimolar 6 after 4 h of stirring at room temperature. Under identical conditions, 81% of (S)-1 and 88% of (S)-5 remained intact.
- T. Kano, M. Ueda, J. Takai and K. Maruoka, J. Am. Chem. Soc., 2006, 128, 6046 CrossRef CAS.
- G. H. Hakimelahi and G. Just, Tetrahedron Lett., 1979, 20, 3645 CrossRef.
- (a) Y. Ye, C. Zheng and R. Fan, Org. Lett., 2009, 11, 3156 CrossRef CAS; (b) R. Fan, H. Wang, Y. Ye and J. Gan, Tetrahedron Lett., 2010, 51, 453 CrossRef CAS.
- M. E. Kuehne and P. J. Reider, J. Org. Chem., 1985, 50, 1464 CrossRef CAS.
- (a) WO, 113337, 2004; (b) WO, 113328, 2004; (c) S. Hu, S. Kelly, S. Lee, J. Tao and E. Flahive, Org. Lett., 2006, 8, 1653 CrossRef CAS.
- WO, 074083, 2003.
- D. Gray and T. Gallagher, Angew. Chem., Int. Ed., 2006, 45, 2419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c1sc00453k |
| This journal is © The Royal Society of Chemistry 2011 |