Use of precatalysts greatly facilitate palladium-catalyzed alkynylations in batch and continuous-flow conditions†
Wei
Shu
and
Stephen L.
Buchwald
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: sbuchwal@mit.edu; Fax: (+1) 617-253-3297
First published on 7th September 2011
Abstract
A general and efficient condition for palladium-catalyzed Heck alkynylation in batch was developed using a novel precatalyst. In addition, the first general and efficient continuous-flow protocol for the coupling of alkynes with aryl bromides in the absence of copper was reported.
Introduction
Palladium-catalyzed carbon–carbon bond-forming reactions play a central role in the construction of key building blocks for many industrially-relevant applications.1 Among these, the palladium-catalyzed cross-coupling of terminal alkynes and aryl halides (Heck alkynylations, also known as Cu-free Sonogashira coupling reactions) can be considered one of the most important transformations, given the synthetic utility of the product aryl alkynes.2 Recently, continuous-flow methods have emerged as interesting new tools for the manufacturing of pharmaceutical compounds or intermediates. Specifically, microfluidic devices allow reactions to be safely performed at high temperatures and pressures, while allowing control of reaction times, obviating the need to handle reactive intermediates, and providing efficient heat- and mass-transfer.3–6While flow-based methods using aryl iodides to facilitate the cross-coupling of sp and sp2carbon atoms have been reported,7,8 these methods suffer in general from relatively high catalyst loadings (>5 mol%) and limited substrate scope. Thus, the development of more versatile processes for palladium-catalyzed alkynylations, specifically under continuous-flow conditions, would be significant. We aimed to develop a method that was also effective with less reactive and expensive electrophiles, such as aryl bromides, chlorides and tosylates. Herein, we report the coupling of alkynes with aryl chlorides and bromides using a palladium precatalyst, which represents the first general and efficient continuous-flow protocol for the coupling of alkynes with aryl bromides in the absence of copper.
Results and discussion
In 2003, our group reported that a catalyst system based on PdCl2(CH3CN)2/XPhos demonstrated excellent reactivity in the Heck alkynylation reaction.9 This system employed 1 mol% of palladium and 3 mol% of XPhos as the ligand. Formation of the catalytically active species was achieved by premixing the ligand and PdCl2(CH3CN)2 for 30 min at room temperature prior to addition of the substrates and base. While this transformation was generally broad in scope, heteroaryl halides and heteroaryl substituted alkynes were less efficiently transformed. We started our investigation by comparing the previously reported conditions with those employing our newly developed second-generation XPhos precatalyst.10 This precatalyst, which was originally developed for Suzuki–Miyaura cross-coupling reactions, can be obtained in a simple one-pot procedure starting from commercially available chemicals, and generates the catalytically active LPd(0) species efficiently upon treatment with a base. With 1 mol% XPhos precatalyst 2a (Fig. 1), we found that full substrate conversion and 86% yield of product could be obtained within 1 h. In contrast, only 74% conversion and 61% yield were obtained under the previously reported conditions (Scheme 1).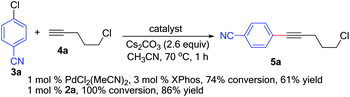 | ||
| Scheme 1 Comparison of two different types of precatalysts. | ||
 | ||
| Fig. 1 Biaryl phosphine ligands and precatalysts. | ||
Next, we investigated the effectiveness of several other precatalysts 2 with various biaryl phosphine ligands (Fig. 2). With a precatalyst based on DavePhos, full conversion was observed after 1 h at 70 °C, affording 5a in 81% yield. When precatalysts with Cy-JohnPhos, SPhos or RuPhos as ligands were employed, only partial conversion was observed. Adding additional XPhos to 2a increased the overall conversion and yield (Fig. 3). Increasing the amount of additional ligand from 0.1 to 1.0 mol% increased the conversion from 36 to 82% at room temperature over a 24 h reaction time. Further increasing the amount of ligand did not offer any additional advantage. Thus, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2a:XPhos was chosen as optimal for further studies. With this combination, most substrate combinations could be successfully converted to product using only 0.25 mol% palladium.
:1 mixture of 2a:XPhos was chosen as optimal for further studies. With this combination, most substrate combinations could be successfully converted to product using only 0.25 mol% palladium.
 | ||
| Fig. 2 Precatalysts 2 with different biaryl phosphine ligands. | ||
 | ||
| Fig. 3 Effect of extra XPhos on the Pd-catalyzed alkynylation reaction. | ||
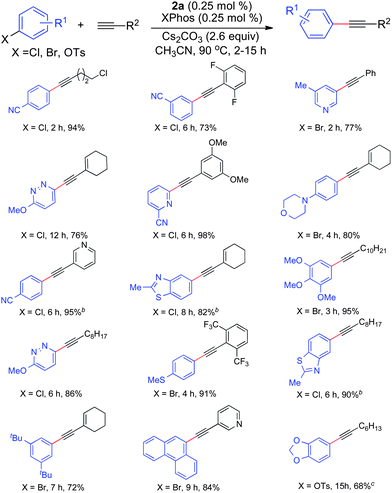
With these optimized conditions in hand, we next examined a variety of substituted aryl halides and pseudo-halides with various alkynes (Table 1). In general, this protocol was effective for a wide range of substrates and manifested broad functional group tolerance. Aryl bromides and chlorides with both electron-donating and electron-withdrawing substituents were all suitable substrates for this transformation. Aryl halides bearing ortho substituents could also be efficiently coupled. Heteroaromatic halides, such as pyridinyl, isoquinolinyl, pyridazinyl, benzothiazolyl and pyrimidinyl, underwent the coupling reaction smoothly, affording the corresponding products in good to excellent yields. In addition, various alkynes with alkyl, aryl and heteroaryl substitutents were efficient coupling partners. It is worth noting that the conditions could be applied to the coupling of an aryl tosylate, which are typically less reactive in alkyne coupling reactions.9,11 Using our previously described conditions, this transformation was only successful when the alkyne was added slowly over the course of the reaction, to diminish alkyne oligomerization.9
|
a
Reaction conditions: The reaction was conducted with ArX (1.0 mmol) and alkyne (1.3 mmol) in 1 mL of acetonitrile at 90 °C for the indicated time; isolated yield based on the average of two runs.
b
2a (1.0 mol%) and XPhos 1a (1.0 mol%) were used. cPrecatalyst 2a (2.0 mol%) and XPhos 1a (2.0 mol%) and alkyne (1.5 mmol) were employed. |
|---|
|
Having demonstrated a broad substrate scope for the alkyne coupling reaction as a batch process, we next focused on applying this reaction protocol to continuous-flow conditions. To be applicable, two key points would be addressed. First, the reaction time should be significantly decreased to minimize the required reactor volume. Second, the use of insoluble bases and the formation of precipitates had to be minimized to prevent clogging of the microchannels. With these in mind, we re-examined different bases for the coupling of 4-bromotoluene with 1-dodecyne (Table 2). We found that when potassium hydroxide was used as the base, full conversion was obtained in only 20 min, affording the desired product in 76% yield (Table 2). With other bases, only partial conversion was observed within the same period of time.
|
|
|||
|---|---|---|---|
| Entry | Base | Conv. (%) | Yield (%) |
| a 0.5 mmol 4-bromotoluene and 0.65 mmol 1-dodecyne were used. GC yield. | |||
| 1 | Cs2CO3 | 54 | 32 |
| 2 | NaOH | 69 | 36 |
| 3 | KOH | 100 | 76 |
| 4 | CsOH | 49 | 43 |

Realizing that the presence of inorganic bases and the formation of bromide precipitates could be problematic for a continuous-flow process, we examined the effect of adding water, that should help dissolve any inorganic salts. When the alkynyl coupling was conducted in a 10:1 mixture of CH3CN–H2O at 100 °C, it reached completion in 10 min. However, we found that precatalyst 2a was only partially soluble in acetonitrile at room temperature. To increase the solubility of the precatalyst, we added DMF as a co-solvent, which afforded the product in 95% GC yield in 10 min. Unfortunately, when other substrates were tested under these conditions, the desired product was obtained along with semi- and fully-hydrogenated products. We hypothesize that DMF serves as a hydrogen donor under these strongly basic conditions.12 Next, we examined alternative solvents and found that the reaction could be completed in dioxane and DMSO, with 66% and 64% yield, respectively. In other solvent systems, full conversion was not observed in 10 min (Table 3). The optimal ratio of dioxane to water was found to be 4:3, producing the product in 100% GC yield. Further dilution of the base had no significant effect on the results (Fig. 4).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Conv. (%) | Yield (%) |
| a 0.5 mmol 4-bromotoluene and 0.65 mmol 1-dodecyne were used. GC yield. | |||
| 1 | Dioxane | 100 | 66 |
| 2 | Dibutyl ether | 89 | 39 |
| 3 | Toluene | 67 | 28 |
| 4 | 1,2-DCE | 59 | 27 |
| 5 | DMSO | 100 | 64 |
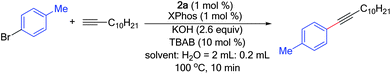
 | ||
| Fig. 4 Biphasic conditions for the Pd-catalyzed alkynylation reaction. | ||
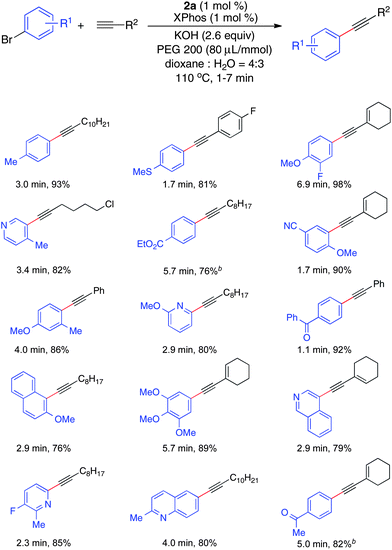
A microfluidic system was assembled as shown in Fig. 5 and 4:3 dioxane–water was employed. Solutions of aryl bromides, alkynes and PEG 200 in dioxane, as well as aqueous potassium hydroxide (0.87 M), were loaded into syringes and introduced into the microfluidic system via one syringe pump. The reagent stream, the base stream, and the solution of XPhos precatalyst 2a and XPhos in dioxane were combined in a cross-mixer. The combined biphasic solution was introduced into a 450 μL packed-bed reactor5a,13 filled with stainless steel spheres at 110 °C with a 10 psi back-pressure regulator. Upon exiting the reactor, the product stream was quenched with a stream of water and ethyl acetate.
 | ||
| Fig. 5 Continuous-flow setup for palladium-catalyzed alkynylations. | ||
This continuous-flow protocol could be applied to the cross-coupling of various aryl bromides and alkynes (Table 4). As in batch, aryl bromides with electron-donating or electron-withdrawing substituents were all good substrates for this transformation. In addition, aryl bromides bearing ortho substituents as well as heteroaromatic bromides were good substrates in this reaction. In the case of base-sensitive substrates containing ethyl ester or acetyl substituents, K3PO4 could be used in place of KOH.
Conclusions
In summary, we have developed a general process for the palladium-catalyzed reaction of aryl chlorides, bromides and tosylates with terminal alkynes in the absence of copper. This new protocol features low catalyst loadings, operationally simple procedures, and a good level functional group tolerance. We have also developed the first general and efficient continuous-flow protocol for the coupling of alkynes with aryl bromides in the absence of copper. This copper-free flow strategy proceeded smoothly with a low palladium catalyst loading and a broad substrate scope. The flow protocol is highly efficient (1–7 min residence time), and provides an attractive and practical way to prepare aryl alkynes on a large scale.Acknowledgements
We thank Novartis International AG for funding.Notes and references
- For recent reviews and books of palladium catalyzed coupling reaction, see: (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) J. P. Wolfe, S. Wagaw, J. F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805 CrossRef CAS; (c) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS; (d) R. F. Heck, Palladium Reagents in Organic Synthesis, Academic Press, London, 1985 Search PubMed; (e) K. Sonogashira, in Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998 Search PubMed; (f) J. A. Marsden and M. M. Haley, in Metal-Catalyzed Cross-Coupling Reactions, 2nd edn, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004 Search PubMed.
- For reviews on Sonogashira coupling, see: (a) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS; (b) H. Doucet and J.-C. Hierso, Angew. Chem., 2007, 119, 850 CrossRef; H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834 Search PubMed; (c) R. R. Tykwinski, Angew. Chem., 2003, 115, 1604 CrossRef; R. R. Tykwinski, Angew. Chem., Int. Ed., 2003, 42, 1566 Search PubMed; (d) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979 CrossRef CAS; (e) R. Rossi, A. Carpita and F. Bellina, Org. Prep. Proced. Int., 1995, 27, 127 CrossRef CAS.
- For reviews on flow chemistry, see: (a) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583 RSC; (b) C. Wiles and P. Watts, Chem. Commun., 2011, 47, 6512 RSC; (c) D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675 RSC; (d) C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687 RSC; (e) A. Cukalovic, J. C. M. R. Monbaliu and C. V. Stevens, Top. Heterocycl. Chem., 2010, 23, 161 CrossRef CAS; (f) R. L. Hartman and K. F. Jensen, Lab Chip, 2009, 9, 2495 RSC; (g) K. Geyer, T. Gustafsson and P. H. Seeberger, Synlett, 2009, 2382 CAS; (h) J.-i. Yoshida, A. Nagaki and T. Yamada, Chem.–Eur. J., 2008, 14, 7450 CrossRef CAS; (i) T. Fukuyama, M. T. Rahman, M. Sata and I. Ryu, Synlett, 2008, 151 CAS; (j) B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. MaQuade, Chem. Rev., 2007, 107, 2300 CrossRef CAS; (k) A. Kirschning, W. Solodenko and K. Mennecke, Chem.–Eur. J., 2006, 12, 5972 CrossRef CAS; (l) K. Geyer, J. D. C. Codée and P. H. Seeberger, Chem.–Eur. J., 2006, 12, 8434 CrossRef CAS; (m) K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., 2004, 116, 410 CrossRef; K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406 Search PubMed.
- For books on microreactors, see: (a) Y.-i. Yoshida, Flash Chemistry: Fast Organic Synthesis in Microsystems, Wiley-Blackwell, Hoboken, 2008 Search PubMed; (b) T. Wirth, Microreactors in Organic Synthesis and Catalysis, Wiley-VCH, Weiheim, 2008 Search PubMed; (c) P. H. Seeberger and T. Blume, New Avenues to Efficient Chemical Synthesis-Emerging Technologies, Springer, Berlin, 2007 Search PubMed; (d) W. Ehrfeld, V. Hessel and H. Löwe, Microreactors: New Technology for Modern Chemistry, Wiley-VCH, Weiheim, 2000 Search PubMed.
- For some selected papers about cross-coupling reactions in flow, see: (a) T. Noël, S. Kuhn, A. J. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., 2011, 123, 6065 CrossRef; T. Noël, S. Kuhn, A. J. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 5943 Search PubMed; (b) T. Noël, J. R. Naber, R. L. Hartman, J. R. McMullen, K. F. Jensen and S. L. Buchwald, Chem. Sci., 2011, 2, 287 RSC; (c) R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2010, 14, 1347 CrossRef CAS; (d) J. P. McMullen, M. T. Stone, S. L. Buchwald and K. F. Jensen, Angew. Chem., 2010, 122, 7230 CrossRef; J. P. McMullen, M. T. Stone, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 7076 Search PubMed; (e) T. N. Glasnov and C. O. Kappe, Adv. Synth. Catal., 2010, 352, 3089 CrossRef CAS; (f) A. Nagaki, A. Kenmoku, U. Moriwaki, A. Hayashi and J.-i. Yoshida, Angew. Chem., 2010, 122, 7705 CrossRef; A. Nagaki, A. Kenmoku, U. Moriwaki, A. Hayashi and J.-i. Yoshida, Angew. Chem., Int. Ed., 2010, 49, 7543 Search PubMed; (g) S. Ceylan, C. Friese, C. Lammel, K. Mazac and A. Kirschning, Angew. Chem., 2008, 120, 9083 CrossRef; S. Ceylan, C. Friese, C. Lammel, K. Mazac and A. Kirschning, Angew. Chem., Int. Ed., 2008, 47, 8950 Search PubMed; (h) H. Kawanami, K. Matsushima, M. Sato and Y. Ikushima, Angew. Chem., 2007, 119, 5221 CrossRef; H. Kawanami, K. Matsushima, M. Sato and Y. Ikushima, Angew. Chem., Int. Ed., 2007, 46, 5129 Search PubMed; (i) E. Vickerstaffe, A.-L. Villard, M. Ladlow and S. V. Ley, Synlett, 2007, 1251 CAS; (j) Y. Uozumi, Y. M. A. Yamada, T. Beppu, N. Fukuyama, M. Ueno and T. Kitamori, J. Am. Chem. Soc., 2006, 128, 15994 CrossRef CAS; (k) G. Shore, S. Morin and M. G. Organ, Angew. Chem., 2006, 118, 2827 CrossRef; G. Shore, S. Morin and M. G. Organ, Angew. Chem., Int. Ed., 2006, 45, 2761 Search PubMed; (l) I. R. Baxendale, C. M. Griffiths-Jones, S. V. Ley and G. K. Tranmer, Chem.–Eur. J., 2006, 12, 4407 CrossRef CAS.
- For a review on cross-coupling in flow, see: T. Noël and S. L. Buchwald, Chem. Soc. Rev., 2011 10.1039/C1CS15075H.
- (a) Y. Zhang, T. F. Jamison, S. Patel and N. Mainolfi, Org. Lett., 2011, 13, 280 CrossRef CAS; (b) A. Sugimoto, T. Fukuyama, M. T. Rahman and I. Ryu, Tetrahedron Lett., 2009, 50, 6364 CrossRef CAS; (c) J. Hillerich and H. Plenio, Chem. Commun., 2003, 3024 RSC; (d) T. Fukuyama, M. Shinmen, S. Nishitani, M. Sato and I. Ryu, Org. Lett., 2002, 4, 1691 CrossRef CAS.
- For catalyst recycling in Sonogashira coupling, see: A. Köllhofer and H. Plenio, Chem.–Eur. J., 2003, 9, 1416 CrossRef.
- D. Gelman and S. L. Buchwald, Angew. Chem., 2003, 115, 6175 CrossRef CAS; D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993 CrossRef.
- (a) X. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klpars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 6653 CrossRef CAS; (b) T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073 CrossRef CAS.
- (a) Activated vinyl tosylates have been used previously for Sonogashira reaction: X. Fu, S. Zhang, J. Yin and D. P. Schumacher, Tetrahedron Lett., 2002, 43, 6673 CrossRef CAS; (b) P. Y. Choy, W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, Chem.–Eur. J., 2010, 16, 9982 CrossRef CAS.
- A. M. Zawisza and J. Muzart, Tetrahedron Lett., 2007, 48, 6738 CrossRef CAS.
- J. R. Naber and S. L. Buchwald, Angew. Chem., 2010, 122, 9659 CrossRef CAS; J. R. Naber and S. L. Buchwald, Angew. Chem., Int. Ed., 2010, 49, 9469 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods and spectral data. See DOI: 10.1039/c1sc00409c |
| This journal is © The Royal Society of Chemistry 2011 |