Enantioselective rhodium-catalyzed arylation of electron-deficient alkenylarenes†‡
Aakarsh
Saxena
and
Hon Wai
Lam
*
EaStCHEM, School of Chemistry, University of Edinburgh, Joseph Black Building, The King's Buildings, West Mains Road, Edinburgh, EH9 3JJ, United Kingdom. E-mail: h.lam@ed.ac.uk:; Tel: +44-131-650-4831
First published on 8th September 2011
Abstract
β-Substituted alkenyl-para-nitroarenes, an unexplored substrate class for catalytic asymmetric addition reactions, undergo highly enantioselective rhodium-catalyzed arylations with arylboronic acids in the presence of a dibenzylamide-containing chiral diene ligand. One example of the asymmetric arylation of an alkenyl-p-cyano-m-(trifluoromethyl)benzene is also presented.
Catalytic enantioselective additions of organometallic reagents to activated alkenes are an important class of reactions for the production of enantioenriched chiral compounds.1 However, examples of such processes where alkenes are activated by arenes or heteroarenes are uncommon, presumably due to the relatively low levels of activation that (hetero)arenes provide. Given the ubiquitous nature of (hetero)arenes in compounds for applications ranging from biology to materials science, the development of reactions that address this deficiency is highly desirable.
Our group has demonstrated that heteroarenes containing a suitably placed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety are able to activate alkenes2 towards enantioselective copper-catalyzed reductions3 and rhodium-catalyzed arylations,4 while Bernadi, Adamo, and co-workers have developed asymmetric additions of nitroalkanes to 4-nitro-5-styrylisoxazoles.5 For alkenes conjugated to simple arenes (which in general provide only minimal activation), highly reactive organometallic reagents are usually required.6 A number of groups have reported stoichiometric or catalytic enantioselective carbolithiations7,8 of various alkenylarenes9–11 mediated by (–)-sparteine9,10 or a (+)-sparteine surrogate.11 Although these carbolithiations work well, the development of processes that proceed under milder conditions, employing organometallic reagents that exhibit greater functional group compatibility, represents an unmet need. In this paper, the catalytic enantioselective addition of arylboronic acids to alkenes conjugated to electron-deficient arenes is described.
N moiety are able to activate alkenes2 towards enantioselective copper-catalyzed reductions3 and rhodium-catalyzed arylations,4 while Bernadi, Adamo, and co-workers have developed asymmetric additions of nitroalkanes to 4-nitro-5-styrylisoxazoles.5 For alkenes conjugated to simple arenes (which in general provide only minimal activation), highly reactive organometallic reagents are usually required.6 A number of groups have reported stoichiometric or catalytic enantioselective carbolithiations7,8 of various alkenylarenes9–11 mediated by (–)-sparteine9,10 or a (+)-sparteine surrogate.11 Although these carbolithiations work well, the development of processes that proceed under milder conditions, employing organometallic reagents that exhibit greater functional group compatibility, represents an unmet need. In this paper, the catalytic enantioselective addition of arylboronic acids to alkenes conjugated to electron-deficient arenes is described.
The rhodium-catalyzed asymmetric 1,4-addition of arylboron compounds to β-substituted electron-deficient alkenes is now a well-established method for the preparation of chiral compounds.12–14 Since the initial discovery of enones as substrates for these reactions,12 subsequent efforts have extended the scope of the acceptor15 to alkenes conjugated to a range of common electron-withdrawing groups.16–22 More recently, less common activating groups have been employed. In addition to our report on asymmetric arylations of alkenylheteroarenes,4 which builds upon work by Lautens and co-workers describing non-enantioselective additions of boronic acids to vinylazines,23 Sasaki and Hayashi have disclosed the asymmetric arylation of borylalkenes.24 However, rhodium-catalyzed addition of arylboron compounds to alkenylarenes has not, to our knowledge, been described. Instead, simple styrenes (where activation of the alkene is minimal) were shown by the Lautens group to undergo Heck-type reactions under aqueous conditions using water-soluble phosphine ligands (eqn (1)).23
 | (1) |
 | (2) |
It occurred to us that placement of the strongly electron-withdrawing nitro group at the para-position of the arene might lead to sufficient polarization of the alkene to the point where addition products 2, rather than Heck-type products, would form, even for 1,2-disubstituted alkenes (eqn (2)). Although nitroalkenes have been successfully employed in myriad additions of carbon nucleophiles,19,25 the analogous reactions of their phenylogous counterparts 1 are extremely rare,6c–h and no asymmetric reactions have been reported. Therefore, the successful realization of the reactions depicted in eqn (2) was an attractive goal and would set the stage for the use of this under-exploited class of electrophiles in other catalytic enantioselective addition reactions.
Our initial experiments focused upon alkenyl-p-nitroarene 1a as a test substrate (Table 1). As a preliminary gauge of reactivity, the addition of PhB(OH)2 to 1a was performed using [Rh(cod)Cl]2 (2.5 mol%) and KOH (2.5 equiv.) in dioxane/H2O at 80 °C under microwave (μw) irradiation26 for 30 min. This experiment resulted in 42% conversion into rac-2a (entry 1). Next, the use of chiral ligands was evaluated in combination with [Rh(C2H4)2Cl]2 as a precatalyst to assess whether 2a could be obtained with improved conversions and in high enantioselectivity. Chiral diene ligands have been shown to provide excellent results in asymmetric 1,4-arylation reactions27–29 and, in view of the success obtained with secondary amide-containing ligand L130 in our study of the asymmetric arylation of alkenylheteroarenes,4 this diene was evaluated first. Although L1 did lead to 2a in 97% ee, the conversion was only 35% (entry 2). Increasing the temperature to 120 °C did increase the conversion with only a slight impact upon enantioselection (95% ee), but appreciable starting material remained (entry 3). Additional amide-containing chiral dienes were then investigated. The enantioselectivity remained high with ligand L2 that lacks the pyrrole on the cyclohexyl ring, but the conversion was low (entry 4). Ligand L34 containing a morpholine amide provided improved conversion (76%) at 80 °C, but the product was formed in only 70% ee (entry 5). Ligands L4 and L5 containing tertiary amides gave improved results (entries 6 and 7), with dibenzylamide-containing ligand L5 giving the product in >95% conversion, 92% isolated yield, and 95% ee (entry 7). In contrast, ligand L6 containing only one benzyl group on the amide nitrogen atom afforded inferior results (entry 8), further suggesting that under these conditions, a tertiary amide in the ligand is beneficial for high conversion. Finally, (R)-BINAP (L7) was tested for comparison and although the enantioselectivity was high, the reaction did not go to completion (entry 9). On the basis of these results, ligand L5 was selected for further study.

a Reactions were conducted using 0.20 mmol of 1a in dioxane (0.5 mL) and H2O (0.1 mL).
b
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Determined by 1H NMR analysis of the unpurified reaction mixtures. cDetermined by HPLC analysis on a chiral stationary phase. d[Rh(cod)Cl]2 was used in place of [Rh(C2H4)2Cl]2, without an additional chiral ligand. eReaction conducted at 120 °C for 30 min. fProduct 2a was isolated in 92% yield. Determined by 1H NMR analysis of the unpurified reaction mixtures. cDetermined by HPLC analysis on a chiral stationary phase. d[Rh(cod)Cl]2 was used in place of [Rh(C2H4)2Cl]2, without an additional chiral ligand. eReaction conducted at 120 °C for 30 min. fProduct 2a was isolated in 92% yield. |
|---|
|
Next, the addition of a range of arylboronic acids to various alkenyl-p-nitroarenes was investigated (Table 2), and the enantioselectivity of the reaction was, in most cases, high (84–97% ee). In addition to a p-nitrophenyl group (entries 1–12), other arenes that provide effective activation in this process include o-fluoro-p-nitrophenyl (entry 14), m-methyl-p-nitrophenyl (entry 15), m-carbomethoxy-p-nitrophenyl (entry 16), and p-nitro-m-(trifluoromethyl)phenyl (entry 17). The reaction is not limited to alkenyl-p-nitrobenzenes; substrate 1k containing a 4-nitronaphthyl group also underwent arylation to provide 2r, though the yield and enantioselectivity were somewhat diminished with this sterically more demanding substrate (entry 18). The range of tolerated substituents at the β-position of the alkene include simple linear alkyl groups (entries 1–9 and 14–18), a cyclopropyl group (entry 10), an allyl ether (entry 11) and an allyl amine (entry 12). However, a β-aryl group was found to inhibit the reaction (entry 13). Regarding the scope of the nucleophile, arylboronic acids containing methyl, halogen, or methoxy substituents were competent reaction partners in this process. The reaction of sterically demanding 2-methylphenylboronic acid with substrate 1b provided 2f in 97% ee, though in a modest 61% yield (entry 6). Thermal heating is as effective as microwave heating, as evidenced by a reaction conducted under otherwise identical conditions (entry 1, values in parentheses). Furthermore, thermal heating was employed in the addition of phenylboronic acid to 1b on a 1.0 mmol scale with 1.25 mol% of [Rh(C2H4)2Cl]2 and 3 mol% of L5 at 80 °C for 1 h, which provided 2c in 83% yield and 95% ee (entry 3).

|
a Unless otherwise stated, reactions were conducted using 0.20 mmol of 1a–1k. Cited yields are of isolated material. Enantiomeric excesses were determined by chiral HPLC analysis.
b
Values in parentheses refer to a reaction conducted under thermal heating under otherwise identical conditions. cReaction performed using 1.0 mmol of 1b at 80 °C under thermal heating for 1 h, using 2.5 mol% of Rh and 3 mol% of L5. dReaction time was 1 h. eReaction performed using 1.0 mmol of 1h. |
|---|

|
An additional demonstration of the reaction scope is provided in eqn (3), where substrate 3 containing a β-trimethylsilyl substituent underwent arylation in 57% yield and 91% ee.31
 | (3) |
To further test the utility of this process, a preparative-scale reaction was performed using substrate 4 (5.0 mmol) containing an oxygenated alkyl substituent at the β-position (Scheme 1). This experiment provided 2t in 88% yield and 93% ee. In addition, reduction of the nitro group of 2t, followed by tosylation of the resulting amine 5, provided sulfonamide 6 in 91% yield over two steps, the absolute stereochemistry of which was determined by single crystal X-ray analysis (Fig. 1).‡32 The sense of enantioinduction observed using ligand L5 is consistent with the stereochemical model proposed for previously reported examples of arylation of acyclic electron-deficient alkenes using structurally similar chiral dienes.4,30 In this model, the rhodium–aryl bond is situated trans to the more electron-deficient alkene, and binding of the alkenylnitroarene occurs in a manner that minimizes unfavorable steric interactions (Fig. 2).
 | ||
| Scheme 1 Larger-scale arylation of 4 and subsequent elaboration. | ||
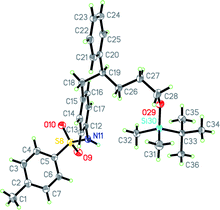 | ||
| Fig. 1 An ORTEP drawing of 6 with ellipsoids set at 50% probability. | ||
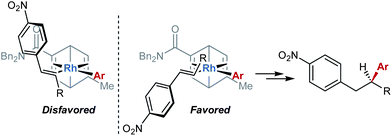 | ||
| Fig. 2 A model for stereochemical induction. | ||
Nitroarenes are well-known to undergo a range of valuable reactions, making them versatile intermediates in the preparation of dyes, pharmaceuticals, and other functional compounds.33 To demonstrate the synthetic utility of the arylation products described herein, 2o was smoothly converted into indole 7 in 67% yield by treatment with vinylmagnesium bromide according to the method of Bartoli and co-workers (eqn (4)).34,35
 | (4) |
Further experiments provided insights into the structural features required in the substrate for the reaction to proceed under the present conditions. Substrates 8 and 9 containing m-nitrophenyl and o-nitrophenyl groups, respectively, did not provide the desired arylation products (Fig. 3).
 | ||
| Fig. 3 Unreactive substrates. | ||
While the lack of reactivity of 8 is not surprising given that the nitro group is not conjugated with the alkene,6d the failure of 9 to undergo arylation was somewhat unexpected, given that o-nitrostyrene has been shown to react smoothly with a variety of active methylene compounds under basic conditions.6d The attempted arylation of 9 using a stoichiometric quantity of the rhodium-ligand complex also provided no evidence of the desired product, suggesting that the problem is one of reactivity rather than catalyst turnover. The addition of 10 mol% of substrate 9 to a repeat of the reaction of Table 2, entry 1 under otherwise identical conditions led to the formation of 2a in >95% conversion and 94% ee, further suggesting that 9 does not poison the catalyst. Exactly how the o-nitro group in 9 inhibits the carborhodation step in the mechanism of rhodium-catalyzed addition of arylboronic acids to electron-deficient alkenes36 is not known at this time.
Nevertheless, the powerful effect of a p-nitro group allowed us to address a problem discovered during our recent study of enantioselective rhodium-catalyzed additions of arylboronic acids to alkenylheteroarenes, which identified a 2-pyridyl group as providing insufficient activation of an adjacent alkene for arylation to proceed efficiently.4 Gratifyingly, 2-alkenylpyridine 10 containing a 5-nitro group underwent arylation in high yield and enantioselectivity (eqn (5)).
 | (5) |
Finally, efforts to employ alkenylbenzene substrates containing a single para-electron-withdrawing substituent other than a nitro group, such as acetyl, nitrile, or methanesulfonyl, were unsuccessful with only low conversions into mixtures of identified products being observed. However, substrate 12, containing a p-cyano-m-(trifluoromethyl)phenyl group, did undergo arylation in 59% yield and 84% ee in the presence of 10 mol% of the rhodium–chiral diene complex after 1.5 h (eqn (6)).
 | (6) |
In contrast, no reaction was observed using (R)-BINAP (L7) as the ligand. The result of eqn (6) suggests that there is scope to increase the range of electron-deficient arenes that can be used as activating groups and future developments in this area may rest upon the identification of more active catalysts and/or improved reaction conditions.
Conclusions
In summary, highly enantioselective rhodium-catalyzed additions of arylboronic acids to alkenyl-p-nitroarenes and an alkenyl-p-cyano-m-(trifluoromethyl)arene have been developed. These reactions represent, to the best of our knowledge, the first examples of catalytic asymmetric additions of air- and moisture-stable organometallic reagents to alkenes activated by electron-deficient arenes. Extension of this concept to other classes of reactions may present exciting new opportunities for asymmetric catalysis. Studies in this area are under way and will be reported in due course.Acknowledgements
This work was supported by a University of Edinburgh MTEM Scholarship to A. S. We thank Benoit Gourdet and Iain D. Roy for assistance in the preparation of ligands, and Dr Fraser J. White for assistance with X-ray crystallography. We are grateful to the EPSRC and the ERC for the award of a Leadership Fellowship and a Starting Grant, respectively, to H. W. L. We also thank the EPSRC National Mass Spectrometry Service Centre at the University of Wales, Swansea, for providing high resolution mass spectra.Notes and references
- For recent reviews, see: (a) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039–1075 RSC; (b) T. Thaler and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 645–648 CrossRef CAS; (c) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS; (d) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 1279–1300 CrossRef CAS; (e) F. Lopez, A. J. Minnaard and B. L. Feringa, Acc. Chem. Res., 2007, 40, 179–188 CrossRef CAS; (f) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS; (g) A. Alexakis and C. Benhaim, Eur. J. Org. Chem., 2002, 3221–3236 CrossRef CAS; (h) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171–196 CrossRef CAS.
- For Ni-catalyzed additions of organometallics to 4-alkenylpyridines proceeding with low (≤15% ee) enantioselectivities, see: I. N. Houpis, J. Lee, I. Dorziotis, A. Molina, B. Reamer, R. P. Volante and P. J. Reider, Tetrahedron, 1998, 54, 1185–1195 CrossRef CAS.
- L. Rupnicki, A. Saxena and H. W. Lam, J. Am. Chem. Soc., 2009, 131, 10386–10387 CrossRef CAS.
- G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373–14375 CrossRef CAS.
- A. Baschieri, L. Bernardi, A. Ricci, S. Suresh and M. F. A. Adamo, Angew. Chem., Int. Ed., 2009, 48, 9342–9345 CrossRef CAS.
- Rare exceptions do exist. For the addition of amides, ketones, imines and nitriles to styrenes, see: (a) H. Pines, S. V. Kannan and J. Simonik, J. Org. Chem., 1971, 36, 2311–2315 CrossRef CAS; (b) A. L. Rodriguez, T. Bunlaksananusorn and P. Knochel, Org. Lett., 2000, 2, 3285–3287 CrossRef CAS. For the intermolecular addition of stabilized carbon nucleophiles to nitrostyrenes, see: (c) H. B. Hass and M. L. Bender, J. Am. Chem. Soc., 1949, 71, 3482–3485 CrossRef CAS; (d) W. J. Dale and C. W. Strobel, J. Am. Chem. Soc., 1954, 76, 6172–6174 CrossRef CAS; (e) J. Wang, B. Chen and J. Bao, J. Org. Chem., 1998, 63, 1853–1862 CrossRef CAS. For the intramolecular addition of stabilized carbon nucleophiles to alkenes conjugated to nitroarenes, see: (f) A. K. Bose, M. S. Manhas and R. M. Ramer, Tetrahedron, 1965, 21, 449–455 CrossRef CAS; (g) D. Craig, M. I. Lansdell and S. E. Lewis, Tetrahedron Lett., 2007, 48, 7861–7864 CrossRef CAS; (h) H. Hu, L.-X. Dai and S.-L. You, Org. Biomol. Chem., 2010, 8, 3207–3210 RSC.
- For early references of alkene carbolithiation, see: (a) P. D. Bartlett, S. Friedman and M. Stiles, J. Am. Chem. Soc., 1953, 75, 1771–1772 CrossRef CAS; (b) P. D. Bartlett, S. J. Tauber and W. P. Weber, J. Am. Chem. Soc., 1969, 91, 6362–6366 CrossRef CAS; (c) P. D. Bartlett, C. V. Goebel and W. P. Weber, J. Am. Chem. Soc., 1969, 91, 7425–7434 CrossRef CAS.
- For reviews, see: (a) A.-M. L. Hogan and D. F. O'Shea, Chem. Commun., 2008, 3839–3851 RSC; (b) J. Clayden, Organolithiums: Selectivity for SynthesisPergamon Press, Oxford, U.K., 2002, pp. 273–335 Search PubMed.
- (a) S. Klein, I. Marek, J.-F. Poisson and J.-F. Normant, J. Am. Chem. Soc., 1995, 117, 8853–8854 CrossRef CAS; (b) S. Norsikian, I. Marek and J.-F. Normant, Tetrahedron Lett., 1997, 38, 7523–7526 CrossRef CAS; (c) S. Norsikian, I. Marek, J.-F. Poisson and J.-F. Normant, Chem.–Eur. J., 1999, 5, 2055–2068 CrossRef CAS; (d) N. Brémand, P. Mangeney and J. F. Normant, Tetrahedron Lett., 2001, 42, 1883–1885 CrossRef.
- (a) A.-M. L. Hogan and D. F. O'Shea, J. Am. Chem. Soc., 2006, 128, 10360–10361 CrossRef CAS; (b) A.-M. L. Hoga and D. F. O'Shea, J. Org. Chem., 2008, 73, 2503–2509 CrossRef; (c) A.-M. L. Hogan, T. Tricotet, A. Meek, S. S. Khokha and D. F. O'Shea, J. Org. Chem., 2008, 73, 6041–6044 CrossRef CAS.
- M. J. Dearden, M. J. McGrath and P. O'Brien, J. Org. Chem., 2004, 69, 5789–5792 CrossRef CAS.
- (a) M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229–4231 CrossRef CAS; (b) Y. Takaya, M. Ogasawara, T. Hayashi, M. Sakai and N. Miyaura, J. Am. Chem. Soc., 1998, 120, 5579–5580 CrossRef CAS.
- For reviews, see ref. 1f and: (a) K. Yoshida and T. Hayashi, In Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH: Weinheim, 2005, chapter 3, p 55–77 Search PubMed; (b) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093–2105 RSC.
- For a review of rhodium-catalyzed carbon–carbon bond-forming reactions of organometallic compounds, see: K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169–196 CrossRef CAS.
- Rhodium-catalyzed additions of arylboron compounds to non-conjugated alkenes is also possible. For additions to strained bicylic alkenes, see: (a) K. Oguma, M. Miura, T. Satoh and M. Nomura, J. Am. Chem. Soc., 2000, 122, 10464–10465 CrossRef CAS; (b) M. Lautens, C. Dockendorff, K. Fagnou and A. Malicki, Org. Lett., 2002, 4, 1311–1314 CrossRef CAS; (c) M. Murakami and H. Igawa, Chem. Commun., 2002, 390–391 RSC; (d) F. Menard and M. Lautens, Angew. Chem., Int. Ed., 2008, 47, 2085–2088 CrossRef CAS; (e) J. Panteleev, F. Menard and M. Lautens, Adv. Synth. Catal., 2008, 350, 2893–2902 CrossRef CAS; (f) J. Bexrud and M. Lautens, Org. Lett., 2010, 12, 3160–3163 CrossRef CAS. For additions to protected allylic amines or allyl sulfones, see: (g) G. C. Tsui, F. Menard and M. Lautens, Org. Lett., 2010, 12, 2456–2459 CrossRef CAS; (h) G. C. Tsu and M. Lautens, Angew. Chem., Int. Ed., 2010, 49, 8938–8941 CrossRef.
- α,β-Unsaturated esters: (a) Y. Takaya, T. Senda, H. Kurushima, M. Ogasawara and T. Hayashi, Tetrahedron: Asymmetry, 1999, 10, 4047–4056 CrossRef CAS; (b) S. Sakuma, M. Sakai, R. Itooka and N. Miyaura, J. Org. Chem., 2000, 65, 5951–5955 CrossRef CAS; (c) J.-F. Paquin, C. R. J. Stephenson, C. Defieber and E. M. Carreira, Org. Lett., 2005, 7, 3821–3824 CrossRef CAS.
- α,β-Unsaturated amides: (a) T. Senda, M. Ogasawara and T. Hayashi, J. Org. Chem., 2001, 66, 6852–6856 CrossRef CAS; (b) S. Sakuma and N. Miyaura, J. Org. Chem., 2001, 66, 8944–8946 CrossRef CAS; (c) R. Shintani, T. Kimura and T. Hayashi, Chem. Commun., 2005, 3213–3214 RSC.
- α,β-Unsaturated aldehydes: J.-F. Paquin, C. Defieber, C. R. J. Stephenson and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 10850–10851 CrossRef CAS.
- Nitroalkenes: (a) T. Hayashi, T. Senda and M. Ogasawara, J. Am. Chem. Soc., 2000, 122, 10716–10717 CrossRef CAS; (b) Z.-Q. Wang, C.-G. Feng, S.-S. Zhang, M.-H. Xu and G.-Q. Lin, Angew. Chem., Int. Ed., 2010, 49, 5780–5783 CrossRef CAS; (c) F. Lang, G. Chen, L. Li, J. Xing, F. Han, L. Cun and J. Liao, Chem.–Eur. J., 2011, 17, 5242–5245 CrossRef CAS.
- α,β-Unsaturated phosphonates: T. Hayashi, T. Senda, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 1999, 121, 11591–11592 CrossRef CAS.
- α,β-Unsaturated sulfones: (a) P. Mauleón and J. C. Carretero, Org. Lett., 2004, 6, 3195–3198 CrossRef; (b) P. Mauleón, I. Alonso, M. R. Rivero and J. C. Carretero, J. Org. Chem., 2007, 72, 9924–9935 CrossRef; (c) P. Mauleón and J. C. Carretero, Chem. Commun., 2005, 4961–4963 RSC.
- Arylmethylene cyanoacetates: S. Sörgel, N. Tokunaga, K. Sasaki, K. Okamoto and T. Hayashi, Org. Lett., 2008, 10, 589–592 CrossRef.
- M. Lautens, A. Roy, K. Fukuoka, K. Fagnou and B. Martín-Matute, J. Am. Chem. Soc., 2001, 123, 5358–5359 CrossRef CAS.
- K. Sasagi and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 8145–8147 CrossRef.
- (a) D. Seebach, E. W. Colvin, F. Lehr and T. Weller, Chimia, 1979, 33, 1–18 CAS; (b) A. G. M. Barrett and G. G. Graboski, Chem. Rev., 1986, 86, 751–762 CrossRef CAS; (c) A. G. M. Barrett, Chem. Soc. Rev., 1991, 20, 95–127 RSC; (d) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877–1894 CrossRef CAS.
- Reactions using microwave heating were carried out in a Biotage microwave synthesizer.
- For a seminal reference, see: T. Hayashi, N. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508–11509 CrossRef CAS.
- For reviews of chiral diene ligands in asymmetric catalysis, see: (a) R. Shintani and T. Hayashi, Aldrichimica Acta, 2009, 42, 31–38 CAS; (b) J. B. Johnson and T. Rovis, Angew. Chem., Int. Ed., 2008, 47, 840–871 CrossRef CAS; (c) C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS.
- For selected recent examples of chiral dienes in catalytic asymmetric 1,4- and 1,6-addition reactions, see ref. 4, 19b and: (a) C.-G. Feng, Z.-Q. Wang, C. Shao, M.-H. Xu and G.-Q. Lin, Org. Lett., 2008, 10, 4101–4104 CrossRef CAS; (b) T. Gendrineau, O. Chuzel, H. Eijsberg, J.-P. Genet and S. Darses, Angew. Chem., Int. Ed., 2008, 47, 7669–7672 CrossRef CAS; (c) X. Hu, M. Zhuang, Z. Cao and H. Du, Org. Lett., 2009, 11, 4744–4747 CrossRef CAS; (d) R. Shintani, Y. Tsutsumi, M. Nagaosa, T. Nishimura and T. Hayashi, J. Am. Chem. Soc., 2009, 131, 13588–13589 CrossRef CAS; (e) M. K. Brown and E. J. Corey, Org. Lett., 2010, 12, 172–175 CrossRef CAS; (f) T. Gendrineau, J.-P. Genet and S. Darses, Org. Lett., 2010, 12, 308–310 CrossRef CAS; (g) X. Hu, Z. Cao, Z. Liu, Y. Wang and H. Du, Adv. Synth. Catal., 2010, 352, 651–655 CrossRef CAS; (h) T. Nishimura, J. Wang, M. Nagaosa, K. Okamoto, R. Shintani, F. Kwong, W. Yu, A. S. C. Chan and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 464–465 CrossRef CAS; (i) Y. Luo and A. J. Carnell, Angew. Chem., Int. Ed., 2010, 49, 2750–2754 CAS; (j) R. Shintani, S. Isobe, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 3795–3798 CrossRef CAS; (k) T. Nishimura, Y. Yasuhara, T. Sawano and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 7872–7873 CrossRef CAS; (l) T. Nishimura, H. Makano, M. Nagaosa and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 12865–12867 CrossRef CAS; (m) R. Shintani and T. Hayashi, Org. Lett., 2011, 13, 350–352 CrossRef CAS; (n) Q. Li, Z. Dong and Z.-X. Yu, Org. Lett., 2011, 13, 1122–1125 CrossRef CAS.
- Ligand L5 is derived from (R)-α-phellandrene. For leading references on the use of (R)-α-phellandrene as a starting material for the construction of chiral dienes, see: (a) K. Okamoto, T. Hayashi and V. H. Rawal, Org. Lett., 2008, 10, 4387–4389 CrossRef CAS; (b) K. Okamoto, T. Hayashi and V. H. Rawal, Chem. Commun., 2009, 4815–4817 RSC.
- For enantioselective Rh-catalyzed additions of organoboron reagents to β-silyl-substituted α,β-unsaturated carbonyl compounds, see ref. 30a and (a) R. Shintani, K. Okamoto and T. Hayashi, Org. Lett., 2005, 7, 4757–4759 CrossRef CAS; (b) R. Shintani, Y. Ichikawa, K. Takatsu, F.-X. Chen and T. Hayashi, J. Org. Chem., 2009, 74, 869–873 CrossRef CAS.
- CCDC 809081 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.
- N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 Search PubMed.
- For the seminal reference, see: G. Bartoli, G. Palmieri, M. Bosco and R. Dalpozzo, Tetrahedron Lett., 1989, 30, 2129–2132 CrossRef CAS.
- For a review of the Bartoli indole synthesis, see: R. Dalpozzo and G. Bartoli, Curr. Org. Chem., 2005, 9, 163–178 CrossRef CAS.
- T. Hayashi, M. Takahishi, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 2002, 124, 5052–5058 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Hamish McNab. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data for new compounds, and crystallographic data. CCDC reference number 809081. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00521a |
| This journal is © The Royal Society of Chemistry 2011 |