Cinchona alkaloid-based phosphoramide catalyzed highly enantioselective Michael addition of unprotected 3-substituted oxindoles to nitroolefins†
Miao
Ding
,
Feng
Zhou
,
Yun-Lin
Liu
,
Cui-Hong
Wang
,
Xiao-Li
Zhao
and
Jian
Zhou
*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, 3663N, Zhongshan Road, Shanghai 200062, China. E-mail: jzhou@chem.ecnu.edu.cn; Fax: (+86) 21-6223-4560
First published on 3rd August 2011
Abstract
A newly developed cinchonidine-derived phosphoramide 6b, simple and easily available, was identified as a powerful catalyst for the highly enantioselective Michael addition of both unprotected 3-aryl and 3-alkyloxindoles to β-substituted nitroalkenes to furnish the C3 quaternary stereogenic carbon center with an adjacent tertiary stereocenter in up to 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity and up to 99% enantioselectivity.
:1 diastereoselectivity and up to 99% enantioselectivity.
Introduction
The catalytic asymmetric construction of full-carbon quaternary stereogenic centers is a challenge in asymmetric catalysis, because of the low reactivity of precursors and additional steric challenges involved in reaction development.1 The use of activating groups to improve the reactivity of substrates is a widely adopted strategy for reaction design. For example, the introduction of an electron-withdrawing group to the nitrogen moiety of 3-substituted oxindoles makes the resulting N-protected oxindoles more reactive towards deprotonative activation.2e As a result, N-Boc protected 3-prochiral oxindole 1 has been much more frequently utilized than unprotected oxindole 2 for the catalytic asymmetric synthesis of 3,3-disubstituted oxindoles,2e which is a privileged heterocyclic motif that constructs the core of many alkaloid natural products, pharmaceutically active compounds and drugs.2 The bulky shielding from the Boc group is also found to be very important for the excellent enantiofacial control in many cases.3 Unfortunately, the introduction and removal of activating groups impose extra cost of time, labor, reagents and energies on the synthesis of target molecules.4 To achieve “ideal synthesis”,4b the use of simple and easily available chiral catalysts and starting materials for the asymmetric catalytic construction of full-carbon stereogenic carbon centers is highly desirable.Recently, the Michael addition of 3-substituted oxindoles 1 to nitroolefins 3 emerged as a versatile method for the synthesis of oxindole and indoline derivatives bearing a quaternary center at the C3 position.5,6 The highly enantioselective conjugate addition of N-Boc protected 3-alkyloxindoles to nitroolefins was pioneered by Barbas III5a and Shibasaki,5b independently. Later, Maruoka found that the high reactivity of N-Boc protected 3-aryloxindoles allowed a novel enantioselective base-free phase-transfer reaction with nitroolefins in a water rich solvent.5c Luo & Cheng reported the addition of N-phenyl 3-methyloxindole and nitroolefins.5d Most recently, Yuan reported the highly enantioselective conjugate addition of N-Boc protected 3-alkyloxindoles to protected 2-amino-1-nitroethenes.5e Despite these achievements, there is still room for further improvement judging by the guidelines of the “ideal synthesis”.4b First, it would be more atom-efficient to use unprotected 3-prochiral oxindoles 2 as the nucleophile, because the synthesis of N-Boc oxindoles 1 from isatins involves three steps, with the sacrifice of one equiv. of (Boc)2O (eqn (1)), and the direct protection of 2 with (Boc)2O was known to be problematic due to the N- and O-selectivity.7
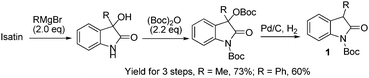 | (1) |
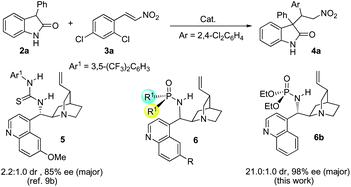
Since Shibata and Toru pioneered the catalytic asymmetric aldol-type reaction of unprotected 3-alkyloxindoles with trifluoropyruvate catalyzed by cinchona alkaloids,8a unprotected 3-prochiral oxindoles have been utilized in the catalytic asymmetric synthesis of 3,3-disubstituted oxindolesvia aldol,8a–c Mannich,8damination8e–g and Michael addition reactions.6b–c However, to the best of our knowledge, most of these protocols were limited to either unprotected 3-aryl or 3-alkyl oxindoles. While a variety of Michael addition reactions of 3-prochiral oxindoles to electron-deficient olefins have been developed,5,6 very limited examples were based on unprotected oxindole 2,6b–c and no method enabled both 3-alkyl and 3-aryloxindoles to work well with β-substituted Michael acceptors in a highly enantioselective manner. Here we wish to report a newly developed phosphoramide 6b as a powerful catalyst for the highly enantioselective Michael addition of unprotected 3-aryl and 3-alkyloxindoles to nitroolefins.
Results and discussions
With our efforts in the synthesis of 3,3-disubstituted oxindoles for biological evaluation,9 we have found that unprotected 3-prochiral oxindoles 2 could readily react with nitroolefins 3 in the presence of Brønsted base catalysts.9b We also tried to develop an asymmetric version of this reaction, but intensive screening of known chiral Brønsted base catalysts only resulted in moderate 2.2:1 dr and 85% ee for the major diastereomer of product 4a, which was obtained by using thiourea catalyst 5.9b In light of this, the development of a new catalyst to improve the diastereo- and enantioselectivity of the Michael addition of unprotected 3-prochiral oxindole 2 to nitroolefin 3 was undertaken in our lab.
Generally, unprotected 3-substituted oxindoles were less reactive nucleophiles, and the pKa value of the C–H bond at the C3 position could be expected to be substantially high, as that of oxindole was 18.2.10 Accordingly, the use of a bifunctional catalyst to activate both the oxindole and nitroalkene might be important for reaction development. Therefore, we designed cinchona alkaloid-derived phosphoramides 6 for the following reasons: 1) amide substituents R1 could serve as shielding groups for two directions, helpful for the stereocontrolled creation of adjacent quaternary and tertiary stereocenters.11 More, it was easy to vary the R1group to tune the steric and electronic properties of the catalysts; 2) the pKa value of the N–H bond of the amide could be easily tuned by varying the R1group from an aryl to an alkoxy group, very useful for the electrophile activation;12 3) these kinds of catalysts were largely unexplored,13 although they could be easily accessed in one step from a cinchona alkaloid-derived primary amine. Only a quinidine-derived phosphinamide catalyst was reported by our group for the enantioselective Strecker reaction of isatin-derived ketoimines using TMSCN.9c
Phosphinamide
6a and phosphoramides 6b–c, obtained in one step from a cinchonidine-derived primary amine and the corresponding phosphinic chloride or chlorophosphates, were first examined in the reaction of unprotected 3-phenyloxindole 2a and nitroalkene 3a in dichloromethane at −10 °C (Table 1). To our delight, all the three catalysts afforded the desired product 4a in excellent ee (entries 1–3). Although phosphinamide 6a could deliver 99% ee for the major diastereomer, the diastereoselectivity was poor (entry 1). Interestingly, phosphoramide 6b gave product 4a in excellent yield, 8:1 dr and 95% ee for the major diastereomer (entry 2). The diphenyl substituted catalyst 6c provided product 4a in slightly lower dr and ee than catalyst 6b (entry 3). These initial results encouraged us to prepare quinine- and cinchonine-derived catalysts 6d and 6e to further improve the selectivity, but both of them were inferior to 6b (entries 4–5). We also synthesized cinchonidine-derived sulfamide catalysts 7a–d14 to compare with catalyst 6b, but they were less diastereo- and enantioselective than catalyst 6b (entries 6–9). On the basis of the above results, the simple and easily available catalyst 6b was chosen for further optimization. Diethyl ether turned out to be the best solvent, and up to 14.0:1.0 dr was achieved for product 4a with 95% ee (entry 10). Lowering the temperature to −20 °C could further improve the dr, but it took four days for the reaction to complete (entry 11). The addition of powdered MS 5 Å could promote the reaction to finish within two days at −20 °C, affording the desired product in 96% yield, with up to 21.0:1.0 dr and 98% ee for the major diastereomer (entry 12).
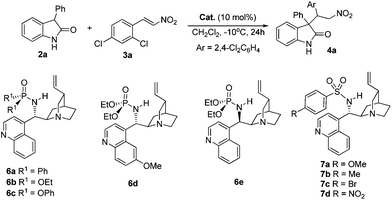
| Entrya | Cat. | Solvent | T /°C | Yieldb (%) | Drc | Ee d (%) (syn/anti) |
|---|---|---|---|---|---|---|
| a Run on a 0.10 mmol scale. b Isolated yield. c Determined by 1H NMR analysis of the crude reaction mixture. d Determined by chiral HPLC analysis. e 1.0 g/1.0 mmol powdered MS 5 Å added. | ||||||
| 1 | 6a | DCM | −10 | 87 | 2.0/1.0 | 99/85 |
| 2 | 6b | DCM | −10 | 96 | 8.0/1.0 | 95/70 |
| 3 | 6c | DCM | −10 | 97 | 6.4/1.0 | 92/72 |
| 4 | 6d | DCM | −10 | 96 | 10.0/1.0 | 87/49 |
| 5 | 6e | DCM | −10 | 76 | 6.0/1.0 | 92/84 |
| 6 | 7a | DCM | −10 | 95 | 8.0/1.0 | 83/70 |
| 7 | 7b | DCM | −10 | 77 | 7.3/1.0 | 81/71 |
| 8 | 7c | DCM | −10 | 95 | 7.0/1.0 | 81/62 |
| 9 | 7d | DCM | −10 | 95 | 7.1/1.0 | 77/64 |
| 10 | 6b | Et2O | −10 | 90 | 14.0/1.0 | 95/66 |
| 11 | 6b | Et2O | −20 | 89 | 18.0/1.0 | 96/71 |
| 12e | 6b | Et2O | −20 | 96 | 21.0/1.0 | 98/73 |
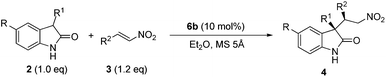
Based on the above results, the substrate scope with respect to both the electrophile and nucleophile was investigated under the optimal conditions: to run the reaction in air, using Et2O as the solvent in the presence of 10 mol% catalyst 6b and MS 5 Å (Table 2). The diastereoselectivity of product 4 was determined by 1H NMR analysis of the crude reaction mixtures, or by HPLC analysis when NMR analysis failed. The reaction of unprotected 3-aryloxindoles with nitroalkenes 3 was first examined. Various nitrostyrene derivatives with electron-rich or -poor substituents on the aromatic ring afforded the desired products 4a–d in excellent ee for the major diastereomer, with high to excellent dr (entries 1–4). Due to the problems in separating the diastereomers and enantiomers by HPLC analysis, (E)-2-(2-nitrovinyl)furan had to work with 5-fluoro-3-phenyloxindole, and product 4e was obtained in excellent ee with moderate dr (entry 5). Aliphatic nitroolefins also worked well at 0 °C to afford the desired products 4f–g in good to high yield and dr, with up to 98% ee for the major diastereomer (entries 6–7). Different 3-aryloxindoles were also examined, and the corresponding products 4h–n were all obtained in high yield and dr, with excellent ee for the major diastereomer (entries 8–14). It should be noted that both the substrate scope and selectivity obtained by phosphoramide 6b in the Michael addition of unprotected 3-aryloxindoles to nitroolefins were obviously better than the literature report which was based on N-Boc protected 3-aryloxindoles.5c
| Entrya | 2 | R2 | 4 | Yieldb(%) | Drc | Ee d (%) |
|---|---|---|---|---|---|---|
| a Run on a 0.25 mmol scale. b Isolated yield. c Determined by 1H NMR analysis of the crude reaction mixture or by HPLC analysis. d Determined by HPLC analysis, for major diastereomer. e −20 °C. f −40 °C. g 0 °C. h Room temperature. i 20 mol% catalyst. | ||||||
| 1e | R = H, R1 = Ph | 2,4-Cl2C6H3 | 4a | 96 | 21:1 |
98 |
| 2e | R = H, R1 = Ph | p-BrC6H4 | 4b | 89 | 13:1 |
99 |
| 3e | R = H, R1 = Ph | p-CF3C6H4 | 4c | 92 | 13:1 |
98 |
| 4e | R = H, R1 = Ph | p-MeOC6H4 | 4d | 85 | 7:1 |
97 |
| 5f | R = F, R1 = Ph | 2-Furanyl | 4e | 84 | 3:1 |
96 |
| 6g | R = H, R1 = Ph | n-Pr | 4f | 76 | 6:1 |
98 |
| 7g | R = H, R1 = Ph | i-Bu | 4g | 84 | 6:1 |
98 |
| 8f | R = F, R1 = Ph | Ph | 4h | 84 | 10:1 |
96 |
| 9f | R = Cl, R1 = Ph | Ph | 4i | 92 | 8:1 |
97 |
| 10f | R = Br, R1 = Ph | Ph | 4j | 78 | 7:1 |
97 |
| 11e | R = OMe, R1 = Ph | Ph | 4k | 83 | 11:1 |
98 |
| 12f | R = H,R1 = p-ClC6H4 | Ph | 4l | 84 | 18:1 |
97 |
| 13e | R = H, R1 = 2-Naphthyl | Ph | 4m | 77 | 11:1 |
99 |
| 14f | R = F, R1 = Ph | p-CF3C6H4 | 4n | 85 | 9:1 |
98 |
| 15h,i |
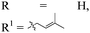
|
p-BrC6H4 | 4o | 78 | 8:1 |
94 |
| 16h,i |
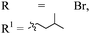
|
p-BrC6H4 | 4p | 89 | 8:1 |
95 |
| 17h,i |
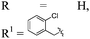
|
p-BrC6H4 | 4q | 95 | 7:1 |
96 |
| 18h,i |
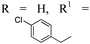
|
p-BrC6H4 | 4r | 95 | 8:1 |
91 |
| 19h,i | R = Br, R1 = Me | p-BrC6H4 | 4s | 77 | 9:1 |
95 |
| 20h,i | R = Br, R1 = Me | 2-Thienyl | 4t | 86 | 12:1 |
95 |
Most importantly, unprotected 3-alkyloxindole turned out to be viable substrates under this reaction condition. Unprotected 3-alkyloxindoles were much less reactive than unprotected 3-aryloxindoles, because their pKa value might be substantially higher than that of oxindole (18.2). As a result, the reaction of unprotected 3-alkyloxindole and nitroolefins was run at room temperature with 20 mol% of catalyst 6b. Because of the problem in separating isomers by HPLC analysis, bromo-substituted nitrostyrene was used as the acceptor, and we were pleased to find that different 3-alkyloxindoles worked well to give the major diastereomer in high diastereoselectivity and excellent enantioselectivity (entries 15–19). Heteroaromatic nitroalkene also worked well with unprotected 3-alkyloxindole to give the major diastereomer of product 4t in high dr and excellent ee (entry 20).
The relative configuration of the major diastereomer of product 4a was confirmed by the X-ray analysis (Fig. 1). The configuration of the stereogenic center of the major diastereomer of product 4m at the C3 position was also assigned to be R and that at the remaining stereocenter was R (see ESI†). Those of other products 4 were tentatively assigned by analogy.
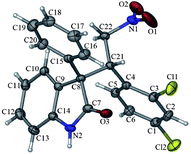 | ||
| Fig. 1 X-ray structure of 4a. | ||
The high diastereo- and enantioselectivity realized by catalyst 6b in this reaction was very impressive. Especially, our method was workable for both unprotected 3-aryl and 3-alkyloxindoles, which was more atom-efficient than literature methods that are based on N-Boc protected 3-prochiral oxindoles. Furthermore, catalyst 6b was very simple and could be easily obtained in only two steps from cheap cinchonidine. These facts made our method very attractive for the synthesis of enantioenriched oxindole or indoline derivatives bearing a quaternary center at the C3 position. These natural product analogues might possess important bioactivity, helpful for drug development.
For example, the reaction of oxindole 2a and nitrostyrene 3h afforded product 4u in 95% yield and 11:1 dr, which could be readily oxidized to acid 8 by a literature method (see Scheme 1).15 The major diastereomer of the oxindole based acid 8 could be isolated in 75% yield with 95% ee, which was an interesting structural motif for medicinal research.2a–g The Michael adduct 4u could also be converted to compound 9 after the reduction and the following cyclization, affording the major diastereomer in 65% yield with 96% ee. The relative and absolute configuration of compound 9 was in accordance with literature report.5c It also should be noted that the isomers of product 4u could not be separated by chiral HPLC analysis, but the ee of compound 8 and 9 was readily determined.
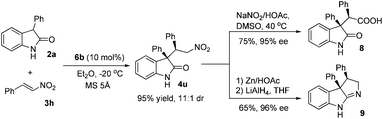 | ||
| Scheme 1 Product elaboration. | ||
Conclusions
In conclusion, we have developed a number of cinchona alkaloid-based phosphoramide catalysts, which are simple and easily available. Among them, cinchonidine-derived bifunctional phosphoramide 6b was identified as a highly diastereo- and enantioselective catalyst for the addition of unprotected 3-prochiral oxindoles to nitroalkenes. Both unprotected 3-aryl and 3-alkyloxindoles could react with a number of different β-substituted nitroalkenes to furnish the C3 quaternary stereogenic carbon center with an adjacent tertiary stereocenter in high diastereo- and enantioselectivity. The modification of the cinchona alkaloid-derived phosphoramides 6 by varying substituents R and R1 to make new bifunctional catalysts, and their application to the catalytic asymmetric construction of all-carbon quaternary stereogenic carbon centers is now under way in our lab.Acknowledgements
The financial support from the 973 program (2011CB808600), National Natural Science Foundation of China (20801018, 20902025), Shanghai Pujiang Program (10PJ1403100), Shanghai Rising-Star Program (10QA1402000), Specialized Research Fund for the Doctoral Program of Higher Education (20090076120007), and the Fundamental Research Funds for the Central Universities (East China Normal University 11043) are highly appreciated.Notes and references
- For reviews, please see: (a) E. J. Corey and A. GuzmanPerez, Angew. Chem., Int. Ed., 1998, 37, 388 CrossRef; (b) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS; (c) I. Denissova and L. Barriault, Tetrahedron, 2003, 59, 10105 CrossRef CAS; (d) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS; (e) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734 CrossRef CAS; (f) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969 CrossRef CAS; (g) M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853 CrossRef CAS; (h) O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873 RSC; (i) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS; (j) J. Chirstoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688 CrossRef CAS.
- For reviews, see: (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS; (b) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209 CrossRef; (c) H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36 CrossRef CAS; (d) B. M. Trost and M. K. Brennan, Synthesis, 2009, 3003 CrossRef CAS; (e) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CAS; (f) S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 Search PubMed; (g) J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Disc., 2010, 13, 758 Search PubMed; for selected examples after the publication of ref. 2e: (h) B. M. Trost and L. C. Czabaniuk, J. Am. Chem. Soc., 2010, 132, 15534 CrossRef CAS; (i) A. P. Antonchick, C. Gerding-Reimers, M. Catarinella, M. Schurmann, H. Preut, S. Ziegler, D. Rauh and H. Waldmann, Nat. Chem., 2010, 2, 735 CrossRef CAS; (j) A. Voituriez, N. Pinto, M. Neel, P. Retailleau and A. Marinetti, Chem.–Eur. J., 2010, 16, 12541 CrossRef CAS; (k) X. Jiang, Y. Cao, Y. Wang, L. Liu, F. Shen and R. Wang, J. Am. Chem. Soc., 2010, 132, 15328 CrossRef CAS; (l) X. Luan, L. Wu, E. Drinkel, R. Mariz, M. Gatti and R. Dorta, Org. Lett., 2010, 12, 1912 CrossRef CAS; (m) X.-N. Wang, Y.-Y. Zhang and S. Ye, Adv. Synth. Catal., 2010, 352, 1892 CrossRef CAS; (n) Q. Wei and L.-Z. Gong, Org. Lett., 2010, 12, 1008 CrossRef CAS; (o) K. Jiang, Z.-J. Jia, S. Chen, L. Wu and Y.-C. Chen, Chem.–Eur. J., 2010, 16, 2852 CrossRef CAS; (p) X. Companyo, A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyanoa and R. Rios, Chem. Commun., 2010, 46, 6953 RSC; (q) P. Chauhan and S. S. Chimni, Chem.–Eur. J., 2010, 16, 7709 CrossRef CAS; (r) Q. Guo, M. Bhanushali and C.-G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 9460 CrossRef CAS; (s) X.-Y. Guang, Y. Wei and M. Shi, Chem.–Eur. J., 2010, 16, 13617 CrossRef; (t) F. Pesciaioli, P. Righi, A. Mazzanti, G. Bartoli and G. Bencivenni, Chem.–Eur. J., 2011, 17, 2842 CrossRef CAS; (u) K. Ohmatsu, M. Kiyokawa and T. Ooi, J. Am. Chem. Soc., 2011, 133, 1307 CrossRef CAS; (v) B. Tan, N. R. Candeias and C. F. BarbasIII, J. Am. Chem. Soc., 2011, 133, 4672 CrossRef CAS; (w) Z.-H. Zhang, W.-H. Zheng and J. C. Antilla, Angew. Chem., Int. Ed., 2011, 50, 1135 CrossRef CAS; (x) L.-L. Wang, L. Peng, J.-F. Bai, L.-N. Jia, X.-Y. Luo, Q.-C. Huang, X.-Y. Xu and L.-X. Wang, Chem. Commun., 2011, 47, 5593 RSC; (y) W.-B. Chen, Z.-J. Wu, J. Hu, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2011, 13, 2472 CrossRef CAS.
- See for examples: (a) Y. Hamashima, T. Suzuki, H. Takano, Y. Shimura and M. Sodeoka, J. Am. Chem. Soc., 2005, 127, 10164 CrossRef CAS; (b) S. Mouri, Z. Chen, H. Mitsunuma, M. Furutachi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 1255 CrossRef CAS; (c) X. Tian, K. Jiang, J. Peng, W. Du and Y.-C. Chen, Org. Lett., 2008, 10, 3583 CrossRef CAS ; also see ref. 6e and 6h.
- (a) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS; (b) P. A. Wender, Chem. Rev., 1996, 96, 1 CrossRef; (c) R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233 CrossRef CAS; (d) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS; (e) I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193 CrossRef CAS; (f) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- For Michael addition of 3-prochiral oxindoles to nitroolefins, see: (a) T. Bui, S. Syed and C. F. Barbas III, J. Am. Chem. Soc., 2009, 131, 8758 CrossRef CAS; (b) Y. Kato, M. Furutachi, Z. Chen, H. Mitsunuma, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 9168 CrossRef CAS; (c) R. He, S. Shirakawa and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 16620 CrossRef CAS; (d) X. Li, B. Zhang, Z.-G. Xi, S. Luo and J.-P. Cheng, Adv. Synth. Catal., 2010, 352, 416 CrossRef CAS; (e) X.-L. Liu, Z.-J. Wu, X.-L. Du, X.-M. Zhang and W.-C. Yuan, J. Org. Chem., 2011, 76, 4008 CrossRef CAS; for reviews on the application of nitroolefins in asymmetric Michael addition, see: (f) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS; (g) A. Alexakis and C. Benhaim, Eur. J. Org. Chem., 2002, 3221 CrossRef CAS; (h) D. Almasi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS; (i) J. L. Vicario, D. Badía and L. Carrillo, Synthesis, 2007, 2065 CrossRef CAS; (j) S. Mukhrjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS; (k) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039 RSC.
- For Michael addition of 3-prochiral oxindoles to other Michael acceptors, see: (a) R. He, C. Ding and K. Maruoka, Angew. Chem., Int. Ed., 2009, 48, 4559 CrossRef CAS; (b) P. Galzerano, G. Bencivenni, F. Pesciaioli, A. Mazzanti, B. Giannichi, L. Sambri, G. Bartoli and P. Melchiorre, Chem.–Eur. J., 2009, 15, 7846 CrossRef CAS; (c) N. Bravo, I. Mon, X. Companyó, A.-N. Alba, A. Moyano and R. Rios, Tetrahedron Lett., 2009, 50, 6624 CrossRef CAS; (d) X. Li, Z.-G. Xi, S. Luo and J.-P. Cheng, Org. Biomol. Chem., 2010, 8, 77 RSC; (e) Q. Zhu and Y. Lu, Angew. Chem., Int. Ed., 2010, 49, 7753 CrossRef CAS; (f) Y.-H. Liao, X.-L. Liu, Z.-J. Wu, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2010, 12, 2896 CrossRef CAS; (g) X. Li, S.-Z. Luo and J.-P. Cheng, Chem.–Eur. J., 2010, 16, 14290 CrossRef CAS; (h) F. Pesciaioli, X. Tian, G. Bencivenni, G. Bartoli and P. Melchiorre, Synlett, 2010, 1704 CAS; (i) W. Sun, L. Hong, C. Liu and R. Wang, Tetrahedron: Asymmetry, 2010, 21, 2493 CrossRef CAS; (j) M. H. Freund and S. B. Tsogoeva, Synlett, 2011, 503 CAS; (k) W. Zheng, Z. Zhang, M. J. Kaplan and J. C. Antilla, J. Am. Chem. Soc., 2011, 133, 3339 CrossRef CAS; (l) S.-W. Duan, J. An, J.-R. Chen and W.-J. Xiao, Org. Lett., 2011, 13, 2290 CrossRef CAS; for reviews on enantioselective Michael addition, see: (m) M. Thirumalaikumar, Organic Preparations and Procedures International, 2011, 43, 67 Search PubMed; (n) H.-C. Guo and J.-A. Ma, Angew. Chem., Int. Ed., 2006, 45, 354 CrossRef CAS; (o) S. Jautze and R. Peters, Synthesis, 2010, 365 CAS; (p) D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058 CrossRef CAS.
- Please see the supporting information of ref. 3a and: B. M. Trost, Y. Zhang and T. Zhang, J. Org. Chem., 2009, 74, 5116 Search PubMed.
- (a) S. Ogawa, N. Shibata, J. Inagaki, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2007, 46, 8666 CrossRef CAS; (b) K. Shen, X.-H. Liu, K. Zheng, W. Li, X.-L. Hu, L.-L. Lin and X.-M. Feng, Chem.–Eur. J., 2010, 15, 3736 CrossRef; (c) K. Shen, X.-H. Liu, W.-T. Wang, G. Wang, W.-D. Cao, W. Li, X.-L. Hu, L.-L. Lin and X.-M. Feng, Chem. Sci., 2010, 1, 590 RSC; (d) L. Cheng, L. Liu, H. Jia, D. Wang and Y.-J. Chen, J. Org. Chem., 2009, 74, 4650 CrossRef CAS; (e) L. Cheng, L. Liu, D. Wang and Y.-J. Chen, Org. Lett., 2009, 11, 3874 CrossRef CAS; (f) Z. Yang, Z. Wang, S. Bai, K. Shen, D. Chen, X. Liu, L. Lin and X. Feng, Chem. Eur. J., 2010, 16, 6632 CAS; (g) K. Shen, X. Liu, G. Wang, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4684 CAS.
- (a) Y.-L. Liu, B.-L. Wang, J.-J. Cao, L. Chen, Y.-X. Zhang, C. Wang and J. Zhou, J. Am. Chem. Soc., 2010, 132, 15176 CrossRef CAS; (b) M. Ding, F. Zhou, Z.-Q. Qian and J. Zhou, Org. Biomol. Chem., 2010, 8, 2912 RSC; (c) Y.-L. Liu, F. Zhou, J.-J. Cao, C.-B. Ji, M. Ding and J. Zhou, Org. Biomol. Chem., 2010, 8, 3847 RSC; (d) Z.-Q. Qian, F. Zhou, T.-P. Du, B.-L. Wang, M. Ding, X.-L. Zhao and J. Zhou, Chem. Commun., 2009, 6753 RSC.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1991, 56, 4218 CrossRef CAS.
- For the use of 3-prochiral oxindoles for the stereocontrolled creation of quaternary at C3 position of oxindole with adjacent tertiary stereocenters, see: (a) X. Tian, K. Jiang, J. Peng, W. Du and Y.-C. Chen, Org. Lett., 2008, 10, 3583 CrossRef CAS; (b) B. M. Trost and Y. Zhang, Chem.–Eur. J., 2010, 16, 296 CrossRef CAS; also see: (c) H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105 CrossRef CAS.
- For reviews on Brønsted acid catalysis, see: (a) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (b) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS; (c) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299 RSC; (d) C. Bolm, T. Rantanen, I. Schiffers and L. Zani, Angew. Chem., Int. Ed., 2005, 44, 1758 CrossRef CAS; (e) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS; (f) Z. Zhan and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC.
- For reviews on the application of cinchona alkaloids derivatives, see: (a) E. M. O. Yeboah, S. O. Yeboah and G. S. Singh, Tetrahedron, 2011, 67, 1725 CrossRef CAS; (b) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229 CrossRef CAS; (c) C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632 RSC; (d) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (e) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS; (f) S. K. Tian, Y. Chen, J. Huang, L. Tang, P. McDaid and L. Deng, Acc. Chem. Res., 2004, 37, 621 CrossRef CAS; (g) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (h) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (i) Y. G. Chen, P. McDaid and L. Deng, Chem. Rev., 2003, 103, 2965 CrossRef CAS.
- (a) A. Peschiulli, B. Procuranti, C. J. O'Connor and S. J. Connon, Nat. Chem., 2010, 2, 380 CrossRef CAS; (b) S. H. Oh, H. S. Rho, J. W. Lee, J. E. Lee, S. H. Youk, J. Chin and C. E. Song, Angew. Chem., Int. Ed., 2008, 47, 7872 CrossRef CAS; (c) J. Luo, L.-W. Xu, R. A. S. Hay and Y. Lu, Org. Lett., 2009, 11, 437 CrossRef CAS; (d) U. C. Döbler, G. M. Mehltretter, W. Baumann and M. Beller, Chirality, 2003, 15, 127 CrossRef CAS.
- C. Matt, A. Wagner and C. Mioskowski, J. Org. Chem., 1997, 62, 234 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 827254. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00390a |
| This journal is © The Royal Society of Chemistry 2011 |