DOI:
10.1039/C1SC00340B
(Edge Article)
Chem. Sci., 2011,
2, 2029-2034
Switching of non-helical overcrowded tetrabenzoheptafulvalene derivatives†
Received
1st June 2011
, Accepted 5th July 2011
First published on 21st July 2011
Abstract
This study explores a series of non-helical overcrowded tetrabenzoheptafulvalene derivatives, which are different from the parent tetrabenzoheptafulvalene (1) by avoiding terminal C–C double bonds, detailing their synthesis, characterization and syn/antiisomerization. The most interesting findings from this study are that the syn and anti isomers of three overcrowded tetrabenzoheptafulvalene derivatives (3–5) can be switched by photo- and thermal isomerizations both in very high yields. The predominance of syn isomer in the photoisomerization has been explained with DFT calculations in terms of the arrangement of the excited- and ground-state potential energy surfaces. The less stable syn isomer of an overcrowded tetrabenzoheptafulvalene derivative thermally isomerizes to the corresponding anti isomer through seven-membered ring flipping, and the energy barrier of thermal isomerization is found to be directly related to the rigidity and crowdedness of the seven-membered rings. These findings suggest that overcrowded tetrabenzoheptafulvalene derivatives are promising building blocks for molecular switches and machines. In addition, the new synthesis and unambiguous characterization of hexabenzoheptafulvalene (4) have also led to correction of the previously claimed synthesis of hexabenzooctalene.
Introduction
Overcrowded alkenes are a representative type of molecular switches based on the photoisomerization of the C–C double bonds.1 Particularly, helical overcrowded alkenes have been widely used as light-driven molecular switches and molecular motors.1,2 However, light-driven switching of non-helical overcrowded alkenes has been rarely explored. As shown in Fig. 1, tetrabenzoheptafulvalene 1 is a symmetric overcrowded bistricyclic ethylene, which exists as syn and anti isomers at room temperature because the conversion between syn and anti conformations through flipping of seven-membered rings is hindered by overcrowding in the fjord regions around the central C–C double bond. When molten at 255 °C or heated in a solution somewhat below 255 °C, the less stable syn isomer undergoes a thermal isomerization to the more stable anti isomer quantitatively3 with an energy barrier of 36.4 kcal mol−1.4 Such thermal isomerization is an interesting feature of this molecule4 in context of thermochromism of overcrowded bistricyclic aromatic ethylenes.5,6 The syn/antiisomerization of 1 in principle can also be triggered by photoisomerization of the central C–C double bond. However, unlike the well known photoisomerization of helical overcrowded alkenes,7syn/antiphotoisomerization of 1 is not observed because as discovered recently,8syn-1 instead undergoes intramolecular [2 + 2] photocycloaddition of terminal C–C double bonds forming a cage-like hydrocarbon, which locks the molecular framework. Here we report an exploratory study into a series of non-helical overcrowded tetrabenzoheptafulvalene derivatives (2–5 in Fig. 1), which do not have two terminal C–C double bonds available for intramolecular [2 + 2] photocycloaddition.9 It is found that 3–5 exist as syn and anti isomers at room temperature, while 2 exists only as the anti isomer at room temperature.4 The anti isomers of 3–5 are converted to the corresponding syn isomers in very high yields upon photoirradiation, while their syn isomers thermally isomerize to the corresponding anti isomers almost quantitatively at elevated temperatures. In contrast, photoirradiation of anti-2 yields not the syn isomer but a ring-closed product. Detailed below are synthesis, characterization and syn/antiisomerization of 2–5 along with a computational study on the photoisomerization and quantitative analysis on the energy barrier for thermal isomerization. This study suggests that non-helical overcrowded tetrabenzoheptafulvalene derivatives are promising building blocks of molecular switches and machines.
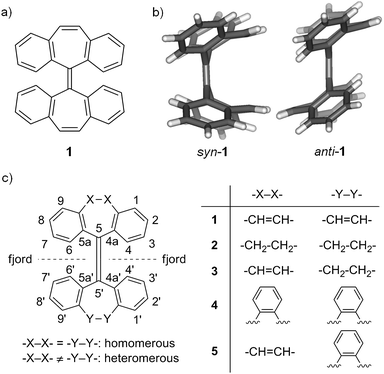 |
| | Fig. 1 (a) Molecular structure of tetrabenzoheptafulvalene (1); (b) structures of syn-1 and anti-1 in the crystals; (c) a general structure for overcrowded tetrabenzoheptafulvalene derivatives (1–5) with numbering. | |
Results and discussion
Synthesis and characterization
As outlined in Scheme 1 and detailed in ESI†, the key for synthesis of 2–5 is a two-step olefin synthesis known as Barton–Kellogg reaction,10 which involves diazo–thioketone coupling followed by desulfurization with a phosphine.11 Unlike the preceding syntheses of 2,3b,12 and 4,13 this method allows synthesis of not only homomerous tetrabenzoheptafulvalene derivatives (2 and 4) but also heteromerous ones (3 and 5). Tetrabenzoheptafulvalenes 4 and 5 were synthesized from thioketone 8 rather than tribenzocycloheptatrien-1-thione because the latter could not be successfully prepared by treating 9H-tribenzo[a,c,e]cyclohepten-9-one14 with Lawesson reagent in our experiments. Desulfuration of episulfides 9 and 10 with triphenylphosphine followed by aromatization under an acidic condition yielded both syn and anti isomers of 4 and 5. In contrast, the Barton–Kellogg reaction yielded only the anti isomers of 2 and 3. This is possibly related to the steric effect of the oxygen-bridged ring.15 Isomer syn-3 was prepared by photoisomerization of anti-3 as shown in Scheme 3.
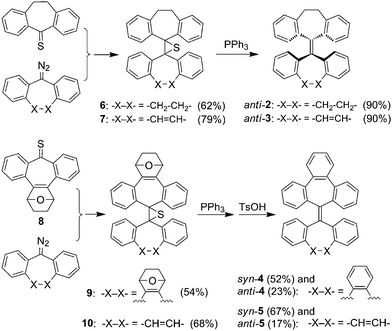 |
| | Scheme 1 Synthesis of 2–5. | |
While the structures of syn-1, anti-1 and anti-2 were identified by X-ray crystallography decades ago,3c,12b the syn/anti structures of 3–5 were first determined based on the 1H NMR spectra and crystal structures in this study. Shown in Fig. 2a is the crystal structure‡ of anti-3 which is characterized by large dihedral angles between the planes of C4–C5–C5′ and C4′–C5′–C5 and between the planes of C6–C5–C5′ and C6′–C5′–C5 (56.3° for both). These dihedral angles are close to those of anti-1 (54.1° for both) and anti-2 (56.1 and 51.7°) as reported by Agranat and Suissa.4 Similar to the two isomers of 1, anti-3 and syn-3 show distinct 1H NMR spectra. Characteristic chemical shifts of the two isomers of 3 are shown in Table 1 together with those of the other tetrabenzoheptafulvalenes. As found from Table 1, the syn isomers of 1, 2 and 3 are characterized by downfield chemical shifts for H4/H6 and H4′/H6′, and the anti isomers of 1 and 3 are characterized by upfield chemical shifts for H4/H6 and H4′/H6′.4 Such conclusion can be extended to other overcrowded tetrabenzoheptafulvalene derivatives, and was applied to 4 and 5 for determining syn and anti structures. The structure of syn-4 was further confirmed with the single-crystal structure‡ shown in Fig. 2b. In addition, the melting points of the anti isomers of 1–5 are generally apparently higher than those of the corresponding syn isomers as shown in Table 1, suggesting that the anti-folded shape allows stronger intermolecular interactions with a tighter molecular packing (the density of anti-1 is 1.6% larger than that of syn-1,3c).
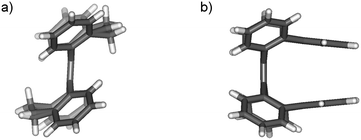 |
| | Fig. 2 Structures of anti-3 and syn-4 in crystals (the vinylene and ethylene groups in anti-3 are disordered). | |
Table 1 Selected chemical shifts (δ) from 1H NMR spectra of 1–5 in CDCl3 and melting points (mp) of 1–5
| |
syn-1 |
anti-1 |
anti-2 |
syn-3 |
anti-3 |
|
δ(H4, H6)/ppm |
7.53 |
6.60 |
6.79 |
7.56 |
6.45 |
|
δ(H4′, H6′)/ppm |
7.53 |
6.60 |
6.79 |
7.42 |
6.90 |
|
δ(alkene H)/ppm |
6.69 |
7.10 |
— |
6.94 |
7.11 |
|
Mp/°C |
255 |
332 |
324 |
231 |
301 |
| |
syn-4 |
anti-4 |
syn-5 |
anti-5 |
|
|
δ(H4, H6)/ppm |
7.59 |
6.52 |
7.46 |
6.60 |
|
|
δ(H4′, H6′)/ppm |
7.59 |
6.52 |
7.49 |
6.53 |
|
|
δ(alkene H)/ppm |
— |
— |
6.02 |
7.08 |
|
|
Mp/°C |
282 |
411 |
249 |
312 |
|
The new synthesis and unambiguous characterization of syn-4 and anti-4 also led to a finding that the previously claimed synthesis of hexabenzooctalene (12)13 was incorrect, as shown in Scheme 2. As reported by Tochtermann in 1963, the reaction of 11 with acid13via Wagner–Meerwein rearrangement yielded a colorless hydrocarbon C38H24 with a melting point of 289–290 °C, and this compound was assigned a structure of hexabenzooctalene (12) as its 1H NMR spectrum (not detailed there) and melting point were not identical to those of hexabenzoheptafulvalene (4). Repeating this reaction indeed led to syn-4 rather than 12, as confirmed with the 1H and 13C NMR spectra as well as the melting point (see ESI† for details). Based on the melting point, the hexabenzoheptafulvalene (4) that was reported in the same literature without characterization should be the anti isomer.
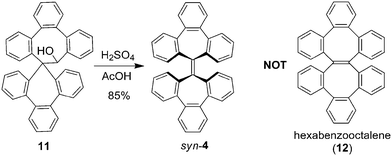 |
| | Scheme 2 Correction of the previously claimed synthesis of hexabenzooctalene (12).13 | |
Isomerization
Photoirradiation of a solution of anti-2 in cyclohexane with a high-pressure mercury lamp for 12 h yielded the ring-closed product 13 (23% as isolated)16 with unreacted anti-2 as found from the 1H NMR spectrum of the crude product (Scheme 3). From the same 1H NMR spectrum, syn-2 was not observed very possibly because syn-2 spontaneously converted to anti-2 through the flipping of seven-membered rings as further discussed in the latter part of this article. As shown in Scheme 3, photoirradiation of a solution of the anti isomers of 3–5 in cyclohexane with a high-pressure mercury lamp generated the corresponding syn isomers in very high yields. It is found that syn-3 gradually converts to anti-3 in a CHCl3 solution within 48 h at room temperature or within 10 min in refluxing benzene, indicating that the thermal isomerization of syn-3 is associated with an energy barrier lower than that of syn-1. This is also in agreement with the finding that the crude product of photoisomerization of anti-3 at room temperature contained 96% of syn-3 and 3% of anti-3. Molten syn-4 and syn-5 converted to the corresponding anti isomers qualitatively within 10 min when heated under an atmosphere of N2 at 400 and 260 °C, respectively. This suggests that the thermal isomerization of syn-5 is associated with an energy barrier similar to that of syn-1 but lower than that of syn-4.
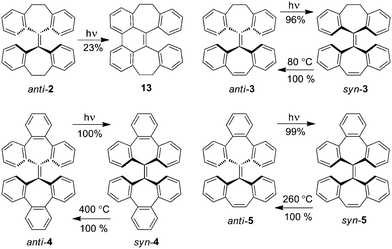 |
| | Scheme 3 Photocyclization of 2 and isomerizations of 3–5. | |
Computational study on photoisomerization
The unusually high yields of syn isomers of 3–5 from photoisomerization of the corresponding anti isomers indicate a photostationary state17 that favors the syn isomer as both the syn and anti isomers are photochemically reactive under UV irradiation. The extensively overlapped absorption spectra of the syn and anti isomers (shown in ESI†) exclude the possibility that only the anti isomer absorbs the UV irradiation thus driving the reaction in one direction. In order to understand why the syn isomers are favored in the photostationary states of 3–5, we studied the reaction coordinate diagrams of both the ground state and the excited state using computational methods.18 Because the photoisomerization involves a half of the molecule rotating around the central C–C double bond, ground-state structures of 3–5 were fully optimized at the Becke3LYP level of density functional theory (DFT)19 with 6-31G* basis set20 with a series of fixed dihedral angles between the planes of C4a–C5–C5a and C4a′–C5′–C5a′ starting from 0° for the anti isomer to 180° for the syn isomer. At each fixed dihedral angle, the energy of singlet excited state was calculated with the optimized geometry of ground state21 using time-dependent density functional theory22 with the same basis set.
Shown in Fig. 3 are the calculated reaction coordinate diagrams for the ground (S0) and excited (S1) states of 3–5 and the proposed reaction pathways with the horizontal axis representing the rotation around the central C–C double bond. It is found that the excited state of 3 has an energy minimum at the rotation angle of 100°,23 while the ground state has two energy maximums at the rotation angles of 70 and 100°. As shown by the blue arrows, when the ground state of either anti or syn isomer is photo-excited to the excited state, the molecule is expected to relax along the S1 potential energy surface to the energy minimum, at which the molecule transits back to its ground state. Then the molecule of 3 can relax to the syn isomer fast in an energy downhill pathway as shown by the red dashed arrow. In contrast, the pathway to the anti isomer (shown by the green dashed arrow) is slowed by an energy barrier of ca. 12 kcal mol−1, which is accompanied by the rotation around the C–C double bond from 80 to 70°. The reaction coordinate diagrams of 4 and 5 are similar and have the energy minimum of the excited state not overlapping with the energy maximum of the ground state. Similar to 3, when 4 and 5 transit from the energy minimum of the excited state back to the ground state, they can relax to the syn isomer fast in an energy downhill pathway as shown by the red dashed arrow. In contrast, the pathway to the anti isomer (shown by the green dashed arrow) is slowed by an energy barrier of ca. 20 kcal mol−1 for 4 and ca. 5 kcal mol−1 for 5. Therefore the unusually high yield of less stable syn isomers of 3–5 from photoisomerization of the corresponding anti isomers can be attributed to the unsymmetrical arrangement of the excited- and ground-state potential energy surfaces. Such unsymmetrical arrangement should be related to the short non-bonding contacts between benzene rings, such as C4⋯C4′, H4⋯H4′ and H4⋯H6′, during rotation around the central C–C double bond.
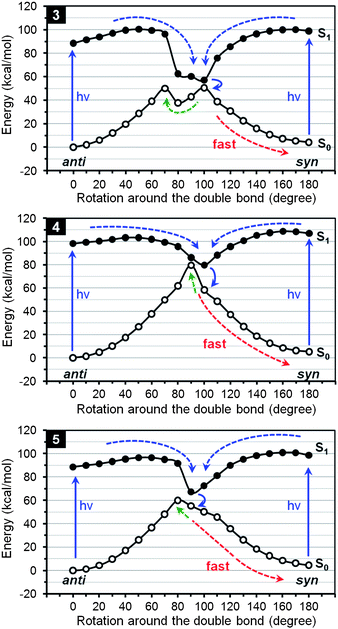 |
| | Fig. 3 Calculated reaction coordinate diagrams for the photoisomerization of 3–5 with proposed reaction pathways. | |
Kinetics of thermal isomerization
Similar to 1,24 overcrowded tetrabenzoheptafulvalenes 2–5 have their anti isomers more stable than the corresponding syn isomers as indicated by the DFT calculations. The calculated energy difference between the anti and syn isomers for 1–5 is in the range of 4.1 kcal mol−1 (for 3) to 5.3 kcal mol−1 (for 4), allowing the anti isomers of 1–5 to predominate at equilibrium. Because the thermal isomerization of the overcrowded tetrabenzoheptafulvalenes is through flipping of the seven-membered rings,4 the observed reactivity in the thermal syn-to-antiisomerization can be understood in terms of the difficulty of seven-membered ring flipping, which is directly related to the rigidity of the seven-membered ring.25 Changing a C–C single bond to a C–C double bond is expected to increase the ring rigidity since the double bond can not rotate as the single bond does. Therefore, the energy barrier for the thermal isomerization of syn-3 is higher than that of syn-2 while lower than that of syn-1. This is in agreement with the fact that syn-3 thermally isomerizes at a lower temperature than syn-1 does. We assume that the energy barrier of 2 is low enough to allow syn-2 to convert to the more stable anti isomer spontaneously at room temperature. This assumption is supported by the fact that syn-2 is not found from the crude products of photochemical reaction of anti-2. The seven-membered rings in 4 and 5 are expected to flip even less readily than those in 1 because the additional benzene rings increase crowdedness by introducing extra short non-bonding contacts and thus further hinder the ring flipping. This is in agreement with that the thermal isomerization of syn-4 occurs at a much higher temperature than syn-1. The fact that the thermal isomerizations of syn-5 and syn-1 occur at similar temperatures suggests that syn-5 thermally isomerizes to anti-5 by flipping the less crowded seven-membered ring, which is the same as that in syn-1.
In order to quantify the above qualitative analysis on the thermal syn-to-antiisomerization, the kinetics of the thermal isomerizations of syn-3 and syn-5 was studied using 1H NMR spectroscopy. The kinetics study did not cover syn-4 because its thermal isomerization requires a temperature too high to be conveniently achieved in a solution.26 The thermal syn-to-antiisomerization is a unimolecular reaction following the rate law of kt = ln([syn]0/[syn]t), where k is the rate constant, t is time, [syn]0 is the initial concentration of syn isomer and [syn]t is the concentration of syn isomer at time t. In the kinetic experiment, a solution of syn-3 in CDCl3 was heated at 40 °C and the progress of syn-to-antiisomerization was monitored with 1H NMR spectroscopy. As shown in Fig. 4a, the methylene protons of syn-3 appear as multiplets at δ = 2.53 and 3.29 ppm, while those of anti-3 appear at δ = 3.02 and 3.71 ppm without overlapping. Therefore the integrations of these peaks were used to calculate the relative concentrations of syn-3 and anti-3, based on which [syn-3]0/[syn-3]t was calculated since [syn-3]0 = [syn-3]t + [anti-3]t. Shown in Fig. 4b is a plot of ln([syn-3]0/[syn-3]t) vs. time, from the slope of which the rate constant k was determined as 2.23 × 10−4 s−1 at 40 °C. Using the Eyring equation k = κ(kBT/h)exp(−ΔG‡/RT) and assuming a value of unity for the transmission coefficient (κ),27 the activation free energy ΔG‡ was calculated as 23.6 kcal mol−1, which is 12.8 kcal mol−1 lower than the reported value of syn-1.4 In a similar manner (see ESI† for details), the thermal isomerization of syn-5 in DMSO-d6 was monitored with 1H NMR spectroscopy, and the rate constant k for thermal isomerization of syn-5 was determined as 4.53 × 10−5s−1 at 186 °C. Using the Eyring equation, the activation free energy ΔG‡ was calculated as 36.4 kcal mol−1, which is the same as the reported value of syn-1, suggesting that syn-5 thermally isomerizes to anti-5 by flipping the less crowded seven-membered ring. As indicated by the above analysis and experimental results, the energy barrier of thermal somerization of overcrowded tetrabenzoheptafulvalenes can be modulated by tuning the rigidity and crowdedness of the seven-membered rings.
![(a) Selected 1H NMR spectra of 3 during the progress of syn-to-antiisomerization in CDCl3 at 40 °C; (b) a plot of ln([syn-3]0/[syn-3]t) vs. time.](/image/article/2011/SC/c1sc00340b/c1sc00340b-f4.gif) |
| | Fig. 4 (a) Selected 1H NMR spectra of 3 during the progress of syn-to-antiisomerization in CDCl3 at 40 °C; (b) a plot of ln([syn-3]0/[syn-3]t) vs. time. | |
Conclusions
In summary, presented above is an exploratory study on syn/anti isomerism of non-helical overcrowded tetrabenzoheptafulvalene derivatives. The most interesting findings in this study are that the syn and anti isomers of 3–5 can be switched by photoisomerization of the central C–C double bond and thermally activated seven-membered ring flipping in very high yields. As indicated by DFT calculations, the predominance of syn isomer in the photoisomerization of 3–5 can be attributed to the arrangement of the excited- and ground-state potential energy surfaces. The kinetics of syn-to-antiisomerization was studied with 1H NMR spectroscopy at elevated temperatures, and the energy barrier of thermal isomerization is found to be related to the rigidity and crowdedness of seven-membered rings. This new family of switchable overcrowded alkenes combine high selectivity in both photo- and thermal isomerizations, tunable energy barrier for thermal isomerization, and relatively large motions of the molecular framework in accompany with isomerization, and thus are promising building blocks for new molecular switches and machines. Moreover, the new synthesis and unambiguous characterization of hexabenzoheptafulvalene (4) have also led to a finding that the previously claimed synthesis of hexabenzooctalene was incorrect.
Acknowledgements
We thank Dr Ken S. M. Yiu (Department of Biology and Chemistry, City University of Hong Kong) for single-crystal crystallography. We thank Dr Chengliang Wang (Department of Electronic Engineering, the Chinese University of Hong Kong) for his help on high-temperature experiments for thermal isomerization. This work was supported by a grant from the Research Grants Council of Hong Kong (project number: GRF402810).
Notes and references
-
Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, Germany, 2001, ch. 5 Search PubMed.
-
(a) B. L. Feringa, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS;
(b) B. L. Feringa, Acc. Chem. Res., 2001, 34, 504–513 CrossRef CAS.
-
(a) A. Schönberg, U. Sodtke and K. Praefcke, Tetrahedron Lett., 1968, 9, 3253–3256 CrossRef;
(b) A. Schönberg, U. Sodtke and K. Praefcke, Chem. Ber., 1969, 102, 1453–1467 CrossRef;
(c) K. S. Dichmann, S. C. Nyburg, F. H. Pickard and J. A. Potworowski, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 27–36 CrossRef CAS.
- I. Agranat and M. R. Suissa, Struct. Chem., 1993, 4, 59–66 CrossRef CAS.
-
(a) P. U. Biedermann, J. J. Stezowski and I. Agranat, Chem.–Eur. J., 2006, 12, 3345–3354 CrossRef CAS;
(b) P. U. Biedermann, J. J. Stezowski and I. Agranat, Eur. J. Org. Chem., 2001, 15–34 CrossRef CAS.
- Compound 1 itself is in fact not thermochromic; see: P. U. Biedermann, I. Agranat and J. J. Stezowski, Chem. Commun., 2001, 954–955 RSC.
-
(a) For recent examples of photo-switching overcrowded alkenes incorporating seven-membered rings, see: W. C. Chen, P. C. Lin, C. H. Chen and C. T. Chen, Chem.–Eur. J., 2010, 16, 12822–12830 CrossRef CAS;
(b) W. C. Chen, Y. W. Lee and C. T. Chen, Org. Lett., 2010, 12, 1472–1475 CrossRef CAS;
(c) Y. Wei, S. Samori, S. Tojo, M. Fujitsuka, J. S. Lin, C. T. Chen and T. Majima, J. Am. Chem. Soc., 2009, 131, 6698–6707 CrossRef CAS.
- M. Pillekamp, W. Alachraf, I. M. Oppel and G. Dyker, J. Org. Chem., 2009, 74, 8355–8358 CrossRef CAS.
- Although 3 and 5 may in principle still undergo an intermolecular [2 + 2] photocyclization, such intermolecular photocyclization should not be able to compete with the intramolecular photoisomerization.
-
(a) D. H. R. Barton and B. J. Willis, Chem. Commun., 1970, 1225–1226 RSC;
(b) R. M. Kellogg and S. Wassenaar, Tetrahedron Lett., 1972, 37, 4045–4060 Search PubMed.
- For recent examples of synthesizing overcrowded tetraaryl ethylenes, see:
(a) N. Assadi, S. Pogodin, S. Cohen, A. Levy and I. Agranat, Struct. Chem., 2009, 20, 541–556 CrossRef CAS;
(b) S. Xiao, M. Myers, Q. Miao, S. Sanaur, K. Pang, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390–7394 CrossRef CAS.
-
(a) I. Agranat, S. Cohen, R. Isaksson, J. Sandström and M. R. Suissa, J. Org. Chem., 1990, 55, 4943–4950 CrossRef CAS;
(b) G. Shoham, S. Cohen, R. M. Suissa and I. Agranat, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1457–1461 CrossRef.
- W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1963, 2, 265–266 CrossRef.
-
(a) S. Martin and A. J. Libbey, Jr, J. Org. Chem., 1957, 22, 1243–1246 CrossRef;
(b) W. Tochtermann, K. Oppenlaender and U. Walter, Chem. Ber., 1964, 97, 1329–1336 CrossRef CAS.
- Acid-catalyzed isomerization of the central C–C double bond is unlikely the reason because treating syn-4 or anti-4 with TsOH did not lead to isomerization in our experiments.
- Molecule 13 can adopt two isomeric structures, namely helical and non-helical isomers. Molecular modeling indicates that the helical isomer is less stable than the non-helical isomer by 0.4 kcal mol−1, and the helical isomer has its inner aromatic H atom close to methylene H atoms with distance shorter than 4.5 Å. 2D ROESY spectrum of 13 showed through-space coupling between the inner aromatic H atom and the methylene H atoms, indicating that it is the helical isomer; see ESI† for details.
-
E. V. Anslyn, D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 2004, ch.16 Search PubMed.
- All the calculations had been performed with the Gaussian 03 software package (M. J. Frisch, et al.., Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, 2004) Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS;
(c) C. Lee, W. Yang and G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
-
(a) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS;
(b) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- For examples of using vertical energy of excited state on the optimized ground-state geometry to simulate the potential energy surface of excited state for alkenes, see:
(a) A. Staykov, J. Areephong, W. R. Browne, B. L. Feringa and K. Yoshizawa, ACS Nano, 2011, 5, 1165 CrossRef CAS;
(b) O. Dmitrenko, W. Reischl, R. D. Bach and J. Spanget-Larsen, J. Phys. Chem. A, 2004, 108, 5662 CrossRef CAS.
-
(a) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS;
(b) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS;
(c) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS;
(d) A. D. Bacon and M. C. Zerner, Theor. Chim. Acta, 1979, 53, 21–54 CrossRef CAS.
- Structural optimization shows that molecule 3 has similar geometries at dihedral angles of 80, 90 and 100° between the planes of C4a–C5–C5a and C4a′–C5′–C5a′. However, rotation from 70 to 80° is associated with a large variation of molecular geometry mainly due to the change in the folding of the seven-membered ring that has five single bonds; see ESI† for optimized geometries.
-
Anti-1 was reported more stable than syn-1 by 19.9 kJ mol−1 as found from calculation at the B3LYP/6-31G* level; see ref. 6.
- M. G. Casarotto and D. J. Craik, J. Pharm. Sci., 2001, 90, 713 CrossRef CAS.
- Refluxing a solution of syn-4 in DMSO-d6 under an atmosphere of N2 for 48 h did not yield anti-4 as observed from 1H NMR spectroscopy.
-
E. V. Anslyn, D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 2004, ch. 7 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, isomerization of 2–5, kinetics study of 3 and 5, energy-optimized geometries of 3–5, and CIF files. CCDC reference numbers 817215 (anti-1) and 817216 (syn-4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00340b |
| ‡ Crystal data for anti-3: C30H22: Mr = 382.48, monoclinic, space groupP21/c, a = 8.5511(3), b = 7.0723(3), c = 16.9235(6) Å, β = 93.189(3)°, V = 1021.88(7) Å3, Z = 2, 3592 reflections collected, 1800 unique (Rint = 0.0120). The final R was 0.045 (all data) and wR was 0.0925 (all data).Crystal data for syn-4: C38H24: Mr = 480.57, monoclinic, space groupP21/c, a = 10.0631(8), b = 15.6693(8), c = 17.3989(12) Å, β = 105.143(8)°, V = 2647.8(3) Å3, Z = 4, 10126 reflections collected, 4691 unique (Rint = 0.0267). The final R was 0.0793 (all data) and wR was 0.1377 (all data). |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.