Straightforward access to aryl-substituted tetrathiafulvalenes by palladium-catalysed direct C–H arylation and their photophysical and electrochemical properties†
Yukihiro
Mitamura
ab,
Hideki
Yorimitsu
*a,
Koichiro
Oshima
*bc and
Atsuhiro
Osuka
a
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: yori@kuchem.kyoto-u.ac.jp; Fax: +81 75 753 3970; Tel: +81 75 753 4010
bDepartment of Material Chemistry, Graduate School of Engineering, Kyoto University, Kyoto-daigaku Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
cEnvironment, Safety and Health Organization, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: oshima@orgrxn.mbox.media.kyoto-u.ac.jp; Fax: +81 75 753 7710; Tel: +81 75 753 2387
First published on 1st August 2011
Abstract
Treatment of tetrathiafulvalene (TTF) with aryl bromides in the presence of cesium carbonate and a palladium catalyst results in direct arylation of TTF. The direct arylation is inherently straightforward and efficient, hence easily creating a large library of aryl TTFs for systematic investigations of structure–property relationships. The present protocol will thus represent an efficient method for exploiting TTF-based interesting functional materials.
Introduction
Tetrathiafulvalene (TTF) undergoes two reversible one-electron oxidations under mild conditions and is well known as an exceptionally good electron donor.1 The charge transfer complexes of TTF, which are composed of its radical cation and the radical anion of an acceptor molecule, tetracyanoquinodimethane (TCNQ) for instance, are notable for their electroconductive, magnetic and photophysical properties and have been leading the progress of molecular electronics.2,3 Not only TTF itself but also peripherally functionalised TTF derivatives are important because they exhibit characteristic properties that stem from their substituents.4 A wide variety of peripherally modified TTFs have hence been synthesised and extensively investigated.1,4Aryl-substituted TTFs seem fascinating because the aromatic substituents can dramatically influence the electronic nature of parent TTF through extension of π-conjugation.5 However, synthetic routes to aryl TTFs are limited virtually to either connecting an aryl-substituted 1,3-dithiole unit with another 1,3-dithiole unit6 or coupling a metalated TTF derivative with an aryl halide under palladium catalysis.7 The former takes multiple steps for overall synthesis and is hence quite laborious. The latter is more sensible since the cross-coupling reactions are nowadays highly reliable tools.8 However, the cross-coupling strategy always requires tedious preparation of metalated TTF intermediates and naturally results in multistep synthesis.7d In particular, the metalation/cross-coupling sequence is quite disadvantageous for the synthesis of densely arylated TTFs. Moreover, functional group compatibility is problematic because the premetalation inevitably provides a lithiated TTF derivative as an intermediate. Development of more efficient approaches to such π-extended aryl TTFs is thereby eagerly awaited.
Results and discussion
Direct arylation of TTF
Transition-metal-catalysed direct C–H arylation of aromatic compounds with aryl halides has been emerging as a powerful alternative to the conventional cross-coupling reaction.9–12 We envisioned that the direct C–H arylation would be applicable to functionalisation of TTF.13 This was indeed the case and, after optimisation of reaction conditions, palladium catalysis has allowed the expected arylation. Treatment of aryl bromides (1 equiv) with TTF (2.5 equiv.) in the presence of cesium carbonate and catalytic amounts of palladium acetate and tri-tert-butylphosphonium tetrafluoroborate in refluxing THF provided the corresponding monoarylated TTFs in good yields (Table 1). The experimental procedure is very simple and thus practical (see Experimental section and ESI†). A variety of functional groups were tolerant under the reaction conditions.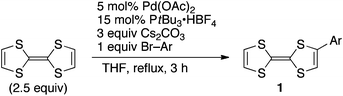
| Ar | 1 | Yield (%) |
|---|---|---|
| a The corresponding three diarylated TTFs (ca. 30% combined yield, determined by 1H NMR) and one triarylated TTF (ca. 5% yield) were formed. | ||
| C6H4-4-Me | 1a | 50a |
| 2-Naphthyl | 1b | 53 |
| C6H4-4-F | 1c | 51 |
| C6H4-4-OMe | 1d | 48 |
| C6H4-4-NMe2 | 1e | 42 |
| C6H4-4-CO2Et | 1f | 64 |
| C6H4-4-NO2 | 1g | 60 |
| C6H4-3-OMe | 1h | 64 |
| C6H4-3-CF3 | 1i | 56 |
| C6H4-4-COMe | 1j | 51 |
| C6H4-4-CN | 1k | 50 |
The exact reaction mechanism for the direct arylation is not clear. It is known that TTF gradually undergoes H/D exchange in the presence of MeONa in MeOD at room temperature to yield TTF-d4.14 The protons of TTF would be acidic enough to be deprotonated by carbonate under the arylation conditions. The high performance of the combination of the palladium catalyst and highly basic cesium carbonate indicates that the reaction would proceed by the concerted metalation–deprotonation pathway.9f,11a,k.l
The modest yields of monoaryl TTFs 1 resulted from competitive di- and triarylation, which, in other words, implies the high efficiency of our strategy. Naturally, we could achieve tetraarylation15–17 of TTF by employing an excess of aryl bromide, a larger amount of the catalyst and prolonged reaction time (Table 2). For the syntheses of 2d, 2e, 2l–2n, 1,4-dioxane was the more suitable solvent because of the higher solubilities of di- and triarylated intermediates in hot dioxane. There are few syntheses of tetraaryl TTFs, which are far less efficient, or employed harsh conditions.18 Our protocol is powerful enough to create a large library of new tetraaryl TTFs with extremely high efficiency.
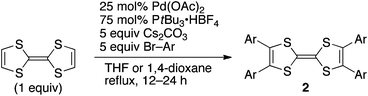
| Ar | Solvent | t/h | 2 | Yield (%) |
|---|---|---|---|---|
| a With 30 mol% Pd(OAc)2, 90 mol% PtBu3·HBF4, 6 equiv. Cs2CO3. | ||||
| C6H4-4-Me | THF | 15 | 2a | 97 |
| 2-Naphthyl | THF | 18 | 2b | 85 |
| C6H4-4-F | THF | 24 | 2c | 60 |
| C6H4-4-OMe | Dioxane | 22 | 2d | 78 |
| C6H4-4-NMe2 | Dioxane | 22 | 2e | 60 |
| C6H4-4-CO2Et | THF | 15 | 2f | 91a |
| C6H4-4-NO2 | THF | 18 | 2g | 60a |
| C6H4-4-CF3 | THF | 16 | 2h | 80a |
| C6H4-3-CF3 | THF | 24 | 2i | 86 |
| C6H4-3-OMe | THF | 24 | 2j | 86 |
| C6H3-3,5-(OMe)2 | THF | 20 | 2k | 82 |
| C6H4-4-CN | Dioxane | 13 | 2l | 60a |
| 3-Pyridyl | Dioxane | 22 | 2m | 63 |
| 4-Biphenylyl | Dioxane | 24 | 2n | 90 |
Structural, photochemical and electrochemical properties of arylated TTFs
The X-ray crystallographic analysis of tetra(4-tolyl)TTF (2a) (Fig. 1) revealed that the dihedral angles between the dithiole unit and the connected tolyl group in the molecule are 36–50°, which promises moderate π-conjugation that permits electronic perturbation by the aryl groups attached. The distances between the two neighbouring TTF mean planes are 3.25 and 4.14 Å and the intermolecular distances between the closest sulfur atoms are 3.93 and 4.20 Å, which shows effective intermolecular electronic communications2–4 through tightly stacked 2a, despite the four tilted 4-tolyl groups.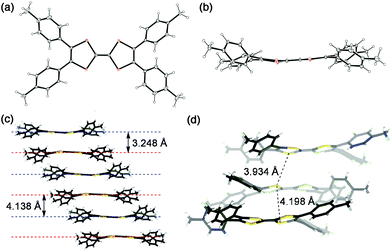 | ||
| Fig. 1 Crystal structure of tetra(4-tolyl)TTF. (a) Top view and (b) side view, (c) stacking structure and (d) close look. | ||
The UV/vis absorption spectra of monoaryl TTFs 1e, 1f and 1g display broad absorption bands in the visible region (Fig. 2a). The absorption bands are dramatically red-shifted as the electron-withdrawing nature of the substituent on the aryl group increase from a dimethylamino group to nitro. The DFT calculations of 1e–1g (Fig. S30, ESI†) revealed several features: (1) the HOMOs are mostly localised on the TTF, (2) the LUMOs are delocalised to the peripheral aryl groups, (3) the visible absorptions correspond to the HOMO–LUMO transitions and (4) the LUMO energies are significantly lowered as the electron-withdrawing nature increase.
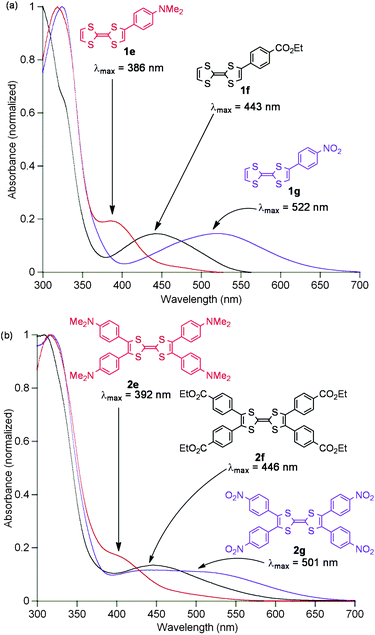 | ||
| Fig. 2 UV/vis spectra of (a) monoaryl TTFs 1e–1g and (b) tetraaryl TTFs 2e–2g in chloroform. | ||
Interestingly, the UV/vis absorption bands of tetraaryl TTFs 2e, 2f and 2g are broader yet similar to those of 1e, 1f and 1g (Fig. 2b). The similarity indicates the HOMO–LUMO energy gaps of aryl TTFs do not heavily depend on the number of the linked aryl groups, as supported by the DFT calculations of 2e–2g (Fig. S32, ESI†).
The electrochemical properties of aryl TTFs 1d–1g and 2d–2g were investigated by cyclic voltammetry (Fig. 3 and Table 3). For both mono- and tetraaryl series, first and second oxidation potentials Eox1 and Eox2 decrease as the aryl substituents are electron-rich, which is consistent with the rises of the HOMO levels of the neutral aryl TTF (Fig. S30 and S32, ESI†) and the SOMO levels of their radical cations (Fig. S31 and S33, ESI†). For instance, tetrakis(4-dimethylaminophenyl)-substituted 2e and its radical cation [2e]˙+ proved to be more easily oxidised by 0.41 V (ΔEox1) and by 0.39 V (ΔEox2) than the corresponding nitro analogues 2g and [2g]˙+, respectively. This arylation could thus readily modulate the oxidation potentials of TTF derivatives. It is worth noting that the substituent-dependent shift of the oxidation potentials for tetraaryl 2e and 2g is much larger than that for monoaryl 1e and 1g (0.13 V for ΔEox1 and 0.14 V for ΔEox2). The oxidation potentials of aryl TTFs were thus found to depend on both the electronic properties and the number of the peripheral aryl groups, which is different from the absorption spectra, that are unrelated to the number of the aryl groups (Fig. 2). Also notably, little changes in the Eox2 − Eox1 values were observed, indicating small differences in Coulombic repulsion between positive charges in the dications for 1d–1g and 2d–2g.
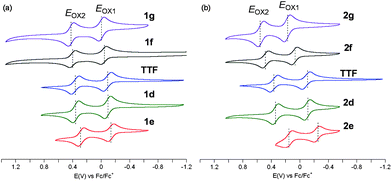 | ||
| Fig. 3 Cyclic voltammograms of (a) monoaryl TTFs 1d–1g and (b) tetraaryl TTFs 2d–2g (TTF for reference). | ||
| Compound | E ox1/V | ΔEox1b/V | E ox2/V | ΔEox2c/V | (Eox2 − Eox1)/V |
|---|---|---|---|---|---|
| a Conditions: 0.1 M Bu4NPF6 in benzonitrile; Ag/Ag+ reference electrode, Pt working electrode and Pt counter electrode. Scan rate 50 mV s−1. V vs. Fc/Fc+ (0.16 V referred to Ag/Ag+). b Defined as Eox1(NO2) − Eox1(CO2Et, OMe or NMe2). c Defined as Eox2(NO2) − Eox2(CO2Et, OMe or NMe2). d For reference. | |||||
| 1g (NO2) | –0.01 | 0.00 | 0.42 | 0.00 | 0.43 |
| 1f (CO2Et) | −0.04 | 0.03 | 0.40 | 0.02 | 0.44 |
| 1d (OMe) | −0.10 | 0.09 | 0.35 | 0.07 | 0.45 |
| 1e (NMe2) | −0.14 | 0.13 | 0.28 | 0.14 | 0.42 |
| 2g (NO2) | 0.17 | 0.00 | 0.54 | 0.00 | 0.37 |
| 2f (CO2Et) | 0.07 | 0.10 | 0.48 | 0.06 | 0.41 |
| 2d (OMe) | −0.11 | 0.28 | 0.34 | 0.20 | 0.45 |
| 2e (NMe2) | −0.24 | 0.41 | 0.15 | 0.39 | 0.39 |
| TTF d | −0.09 | — | 0.36 | — | 0.45 |
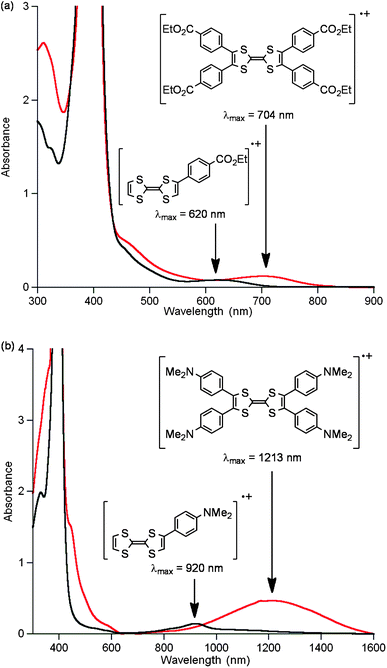
Tetraaryl TTFs 2a, 2d, 2e and 2f reacted with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) to yield the corresponding 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 charge transfer (CT) complexes. The UV/vis/NIR absorption spectra of the CT complexes in acetonitrile showed typical broad absorption bands in the >600 nm region (Fig. 4).2–4 The absorption bands are significantly red-shifted as the electron-donating nature of the aryl group increase, which is opposite to the order for neutral aryl TTFs (Fig. 2). In particular, the NIR absorption of the CT complex of 2e is centred around 1100 nm and reaches 1600 nm.
:1 charge transfer (CT) complexes. The UV/vis/NIR absorption spectra of the CT complexes in acetonitrile showed typical broad absorption bands in the >600 nm region (Fig. 4).2–4 The absorption bands are significantly red-shifted as the electron-donating nature of the aryl group increase, which is opposite to the order for neutral aryl TTFs (Fig. 2). In particular, the NIR absorption of the CT complex of 2e is centred around 1100 nm and reaches 1600 nm.
 | ||
| Fig. 4 UV/vis/NIR absorption spectra of (4-RC6H4)4TTF–DDQ complexes in acetonitrile. | ||
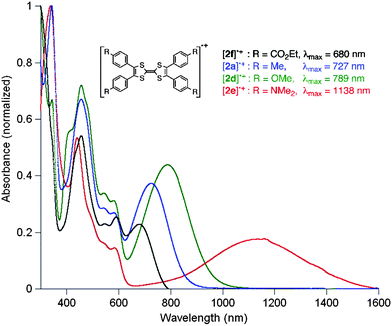
The CT complexes of monoaryl 1e and 1f and tetraaryl 2e and 2f with TCNQ showed UV/vis/NIR absorption spectra as illustrated in Fig. 5. Interestingly, the tetraaryl TTF CT complexes display red-shifted absorptions compared to the corresponding monoaryl ones. The number of the aryl groups in the aryl TTFs, as well as the electronic characters, has a considerable influence on the absorptions of their cation radicals, while being unrelated to those of the neutral aryl TTFs (Fig. 2).
 | ||
| Fig. 5 UV/vis/NIR absorption spectra of aryl TTF–TCNQ complexes in 1,2-dichloroethane: (a) complexes of 1f and 2f; (b) complexes of 1e and 2e. | ||
Experimental†
Typical procedure for palladium-catalysed monoarylation reactions
Pd(OAc)2 (2.3 mg, 0.01 mmol), PtBu3·HBF4 (8.7 mg, 0.03 mmol) and Cs2CO3 (195.5 mg, 0.60 mmol) were placed in a 20-mL reaction flask under nitrogen. THF (0.5 mL) was added and the mixture was stirred for 10 min at reflux. A solution of tetrathiafulvalene (102.2 mg, 0.50 mmol) and 4-bromotoluene (34.2 mg, 0.20 mmol) in THF (0.5 mL) was added. The mixture was heated at reflux for 3 h. The organic compounds were extracted with dichloromethane three times. The combined organic part was washed with brine, passed through a pad of Florisil, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by gel permeation chromatography with chloroform as eluent to afford 2-(4-methylphenyl)tetrathiafulvalene (1a) as a yellow solid (30.0 mg, 0.10 mmol, 50%).Typical procedure for palladium-catalysed tetraarylation reactions
Pd(OAc)2 (8.4 mg, 0.038 mmol), PtBu3·HBF4 (32.6 mg, 0.11 mmol) and Cs2CO3 (244.4 mg, 0.75 mmol) were placed in a 20-mL reaction flask under nitrogen. THF (1.0 mL) was added and the mixture was stirred for 10 min at reflux. A solution of tetrathiafulvalene (30.7 mg, 0.15 mmol) and 4-bromotoluene (128.3 mg, 0.75 mmol) in THF (1.0 mL) was added. The mixture was heated at reflux for 15 h. The organic compounds were extracted with chloroform three times. The combined organic part was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. Chromatographic purification on silica gel by using hexane–dichloromethane (or chloroform) as eluent afforded 2,3,6,7-tetra(4-methylphenyl)tetrathiafulvalene (2a) (84.3 mg, 0.15 mmol, 100%) as a red solid.Crystallographic data for 2a
C34H28S4, Mr = 564.85, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.217(5), b = 11.524(4), c = 15.597(6) Å, α = 100.750(14), β = 105.128(17), γ = 110.659(17)°, V = 1423.6(10) Å3, T = 123 K, Z = 2, R1 = 0.0342 (I > 2σ(I)), wR2 = 0.0948 (all data), GOF = 1.007.
, a = 9.217(5), b = 11.524(4), c = 15.597(6) Å, α = 100.750(14), β = 105.128(17), γ = 110.659(17)°, V = 1423.6(10) Å3, T = 123 K, Z = 2, R1 = 0.0342 (I > 2σ(I)), wR2 = 0.0948 (all data), GOF = 1.007.
Conclusions
We have reported straightforward synthesis of aryl TTFs by using palladium-catalysed direct C–H arylation. Our protocol is highly efficient to create a large library of aryl TTFs. The library has allowed us to systematically investigate the photophysical and electrochemical properties of aryl TTFs and their CT complexes. The present protocol offers a highly efficient tool for exploiting electroconductive TTF-based materials and functional materials of photo- and electrochemical interest.Acknowledgements
This work was supported by Grants-in-Aid (nos. 22245006 (A) and 20108006 “pi-Space” and 22106523 “Reaction Integration”) and GCOE Research from MEXT and JSPS. We thank Professor Hideki Yamochi (Research Center for Low Temperature and Materials Sciences, Kyoto University) for helpful discussion. H. Y. thanks financial support from Kinki Invention Center.Notes and references
- TTF Chemistry, ed. J. Yamada and T. Sugimoto, Kodansha, 2004 Search PubMed.
- Early examples: (a) J. Ferraris, D. O. Cowan, V. Walatka, Jr. and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948 CrossRef CAS; (b) R. C. Wheland, J. Am. Chem. Soc., 1976, 98, 3926 CrossRef CAS; (c) E. M. Engler, B. A. Scott, S. Etemad, T. Penney and V. V. Patel, J. Am. Chem. Soc., 1977, 99, 5909 CrossRef CAS.
- Reviews: (a) G. Saito and Y. Yoshida, Bull. Chem. Soc. Jpn., 2007, 80, 1 CrossRef CAS; (b) D. Jérome, Chem. Rev., 2004, 104, 5565 CrossRef.
- (a) T. Otsubo and K. Takimiya, Bull. Chem. Soc. Jpn., 2004, 77, 43 CrossRef CAS; (b) A. Gorgues, P. Hudhomme and M. Salle, Chem. Rev., 2004, 104, 5151 CrossRef CAS; (c) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS; (d) J. L. Segura and N. Martin, Angew. Chem., Int. Ed, 2001, 40, 1371 CrossRef; (e) J. Yamada and H. Akutsu, Chem. Rev., 2004, 104, 5057 CrossRef CAS; (f) S. Horiuchi, H. Yamochi, G. Saito, K. Sakaguchi and M. Kusunoki, J. Am. Chem. Soc., 1996, 118, 8604 CrossRef CAS; (g) F. Pointillart, T. Cauchy, O. Maury, Y.-L. Gal, S. Golhen, O. Cador and L. Ouahab, Chem.–Eur. J., 2010, 16, 11926 CrossRef CAS.
- (a) H. Fujiwara, Y. Sugishima and K. Tsujimoto, Tetrahedron Lett., 2008, 49, 7200 CrossRef CAS; (b) T. Murata, Y. Morita, Y. Yakiyama, K. Fukui, H. Yamochi, G. Saito and K. Nakatsuji, J. Am. Chem. Soc., 2007, 129, 10837 CrossRef CAS; (c) Y. Wang, S. Cui, B. Li, J. Zhang and Y. Zhang, Cryst. Growth Des., 2009, 9, 3855 CrossRef CAS; (d) J. F. Lamere, I. Malfant, A. S. Saquet and P. G. Lacroix, Chem. Mater., 2007, 19, 805 CrossRef CAS; (e) Q.-Y. Zhu, L.-B. Huo, Y.-R. Qin, Y.-P. Zhang, Z.-J. Lu, J.-P. Wang and J. Dai, J. Phys. Chem. B, 2010, 114, 361 CrossRef CAS; (f) Q.-Y. Zhu, Y. Liu, W. Lu, Y. Zhang, G.-Q. Bian, G.-Y. Niu and J. Dai, Inorg. Chem., 2007, 46, 10065 CrossRef CAS; (g) K. Herve, S.-X. Liu, O. Cador, S. Golhen, Y.-L. Gal, A. Bousseksou, H. Stoeckli-Evans, S. Decurtins and L. Ouahab, Eur. J. Inorg. Chem., 2006, 3498 CrossRef CAS.
- (a) C. U. PittmanJr., M. Narita and Y.-F. Liang, J. Org. Chem., 1976, 41, 2855 CrossRef; (b) M. L. Kaplan, R. C. Haddon, F. Wudl and E. D. Feit, J. Org. Chem., 1978, 43, 4642 CrossRef CAS; (c) C. Polycarpe, E. Torreilles, L. Giral, A. Babeau, N.-H. Tinh and H. Gasparoux, J. Heterocycl. Chem., 1984, 21, 1741 CrossRef CAS.
- (a) M. Iyoda, Y. Kuwatani, N. Ueno and M. Oda, J. Chem. Soc., Chem. Commun., 1992, 153 Search PubMed; (b) S. Bouguessa, A. K. Gouasmia, S. Golhen, L. Ouahab and J. M. Fabre, Tetrahedron Lett., 2003, 44, 9275 CrossRef CAS; (c) L. Wang, B. Zhang and J. P. Zhang, Inorg. Chem., 2006, 45, 6860 CrossRef CAS; (d) M. Iyoda, M. Hasegawa and Y. Miyake, Chem. Rev., 2004, 104, 5085 CrossRef CAS; (e) A. J. Moore, A. S. Batsanov, M. R. Bryce, J. A. K. Howard, V. Khodorkovsky, L. Shapiro and A. Shames, Eur. J. Org. Chem., 2001, 73 CrossRef CAS.
- (a) Cross-Coupling Reactions: A Practical Guide, ed. N. Miyaura, Springer, 2010 Search PubMed; (b) Metal-Catalysed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley, 2004 Search PubMed.
- Reviews: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (b) T. Satoh and M. Miura, Chem. Lett., 2007, 36, 200 CrossRef CAS; (c) A. Mori and A. Sugie, Bull. Chem. Soc. Jpn., 2008, 81, 548 CrossRef CAS; (d) T. Satoh and M. Miura, Synthesis, 2010, 3395 CrossRef CAS; (e) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (f) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef; (g) K. Hirano and M. Miura, Synlett, 2011, 294 CAS; (h) C–H Activation, ed. J.-Q. Yu and Z. Shi, Springer, 2010 Search PubMed; (i) O. Daugulis, V. G. Zaitsev, D. Shabashov, Q.-N. Pham and A. Lazareva, Synlett, 2006, 3382 CrossRef CAS; (j) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC.
- Pioneering works: (a) N. Nakamura, Y. Tajima and K. Sakai, Heterocycles, 1982, 17, 235 CrossRef CAS; (b) Y. Akita, Y. Itagaki, S. Takizawa and A. Ohta, Chem. Pharm. Bull., 1989, 37, 1477 CAS; (c) Y. Aoyagi, A. Inoue, I. Koizumi, R. Hashimoto, K. Tokunaga, K. Gohma, J. Komatsu, K. Sekine, A. Miyafuji, J. Kunoh, R. Homma, Y. Akita and A. Ohta, Heterocycles, 1992, 33, 257 CrossRef CAS; (d) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed, 1997, 36, 1740 CrossRef CAS.
- Very recent representative examples: (a) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS; (b) X. Wang, D. V. Gribkov and D. Sames, J. Org. Chem., 2007, 72, 1476 CrossRef CAS; (c) J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858 CrossRef CAS; (d) K. Kitazawa, T. Kochi, M. Sato and F. Kakiuchi, Org. Lett., 2009, 11, 1951 CrossRef CAS; (e) M. Tobisu, I. Hyodo and N. Chatani, J. Am. Chem. Soc., 2009, 131, 12070 CrossRef CAS; (f) J. Canivet, J. Yamaguchi, I. Ban and K. Itami, Org. Lett., 2009, 11, 1733 CrossRef CAS; (g) A. M. Wagner and M. S. Sanford, Org. Lett., 2011, 13, 288 CrossRef CAS; (h) E. T. Nadres, A. Lazareva and O. Daugulis, J. Org. Chem., 2011, 76, 471 CrossRef CAS; (i) M. J. Tredwell, M. Gulias, N. G. Bremeyer, C. C. C. Johansson, B. S. L. Collins and M. J. Gaunt, Angew. Chem., Int. Ed, 2011, 50, 1076 CrossRef CAS; (j) D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef; (k) D. García-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 6880 Search PubMed; (l) C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2009, 131, 10844 CrossRef CAS.
- Our previous work: (a) M. Iwasaki, H. Yorimitsu and K. Oshima, Chem.–Asian. J., 2007, 2, 1430 CrossRef CAS; (b) Y. Kawamata, S. Tokuji, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2011, 50 DOI:10.1002/anie.201102318.
- Application of C–H arylation to material sciences: (a) A. Mori, A. Sekiguchi, K. Masui, T. Shimada, M. Horie, K. Osakada, M. Kawamoto and T. Ikeda, J. Am. Chem. Soc., 2003, 125, 1700 CrossRef CAS; (b) M. Takahashi, K. Masui, H. Sekiguchi, N. Kobayashi, A. Mori, M. Funahashi and N. Tamaoki, J. Am. Chem. Soc., 2006, 128, 10930 CrossRef CAS; (c) K. Masui, A. Mori, K. Okano, K. Takamura, M. Kinoshita and T. Ikeda, Org. Lett., 2004, 6, 2011 CrossRef CAS; (d) N. Masuda, S. Tanba, A. Sugie, D. Monguchi, N. Koumura, K. Hara and A. Mori, Org. Lett., 2009, 11, 2297 CrossRef CAS; (e) A. Yokooji, T. Satoh, M. Miura and M. Nomura, Tetrahedron, 2004, 60, 6757 CrossRef CAS; (f) M. Miyasaka, A. Fukushima, T. Satoh, K. Hirano and M. Miura, Chem.–Eur. J., 2009, 15, 3674 CrossRef CAS; (g) Q. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420 CrossRef CAS; (h) J. Hassan, E. Schulz, C. Gozzi and M. Lemaire, J. Mol. Catal. A: Chem., 2003, 195, 125 CrossRef CAS; (i) Also see ref. 9c and.12b.
- D. Dolphin, W. Pegg and P. Wirz, Can. J. Chem., 1974, 52, 4078 CrossRef CAS.
- The first example of multiple arylation using the specific catalytic system Pd(OAc)2/PtBu3/Cs2CO3: M. Miura, S. Pivsa-Art, G. Dyker, J. Heiermann, T. Satoh and M. Nomura, Chem. Commun., 1998, 1889 RSC.
- Multiple arylation: (a) F. Shibahara, E. Yamaguchi and T. Murai, Chem. Commun., 2010, 46, 2471 RSC; (b) T. Satoh, M. Miura and M. Nomura, J. Organomet. Chem., 2002, 653, 161 CrossRef CAS; (c) T. Okazawa, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2002, 124, 5286 CrossRef CAS; (d) I. Ban, T. Sudo, T. Tanoguchi and K. Itami, Org. Lett., 2008, 10, 3607 CrossRef CAS; (e) D. Shabashov and O. Daugulis, Org. Lett., 2006, 8, 4947 CrossRef CAS; (f) S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Org. Lett., 2001, 3, 2579 CrossRef CAS; (g) J.-M. Joo, B. B. Toure and D. Sames, J. Org. Chem., 2010, 75, 4911 CrossRef CAS; (h) K. Kitazawa, T. Kochi, M. Sato and F. Kakiuchi, Org. Lett., 2009, 11, 1951 CrossRef CAS; (i) S. Nakazono, S. Easwaramoorthi, D. Kim, H. Shinokubo and A. Osuka, Org. Lett., 2009, 11, 5426 CrossRef CAS; (j) Also see ref. 9d, 10d, 12b, 13f and 13g.
- Reviews on the importance of one-pot multistep syntheses: (a) S. Suga, D. Yamada and J. Yoshida, Chem. Lett., 2010, 39, 404 CrossRef CAS , and references cited therein; (b) A. J. Bard, Integrated Chemical Systems, Wiley: 1994 Search PubMed; (c) Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley, 2005 Search PubMed.
- Lengthy synthesis: A. Charlton, A. E. Underhill, G. Williams, M. Kalaji, P. J. Murphy, D. E. Hibbs, M. B. Hursthouse and K. M. A. Malik, Chem. Commun., 1996, 2423 RSC ; condensation of diarylacetylene with carbon disulfide under 4500 atm: T. Nagawa, Y. Zama and Y. Okamoto, Bull. Chem. Soc. Jpn., 1984, 57, 2035 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures, characterisation data, X-ray crystallographic data, details of DFT calculations. CCDC reference number 818657. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00372k |
| This journal is © The Royal Society of Chemistry 2011 |