Enhanced carbon dioxide capture upon incorporation of N,N′-dimethylethylenediamine in the metal–organic framework CuBTTri†
Thomas M.
McDonald
a,
Deanna M.
D'Alessandro
b,
Rajamani
Krishna
c and
Jeffrey R.
Long
*a
aDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: jrlong@berkeley.edu
bSchool of Chemistry, The University of Sydney, Sydney, New South Wales 2006, Australia
cVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands
First published on 2nd August 2011
Abstract
High capacity, high selectivity, and low-cost regeneration conditions are the most important criteria by which new adsorbents for post-combustion carbon dioxide capture will be judged. The incorporation of N,N′-dimethylethylenediamine (mmen) into H3[(Cu4Cl)3(BTTri)8 (CuBTTri; H3BTTri = 1,3,5-tri(1H-1,2,3-triazol-4-yl)benzene), a water-stable, triazolate-bridged framework, is shown to drastically enhance CO2 adsorption, resulting in one of the best performing metal–organic frameworks for CO2 separation reported to date. High porosity was maintained despite stoichiometric attachment of mmen to the open metal sites of the framework, resulting in a BET surface area of 870 m2 g−1. At 25 °C under a 0.15 bar CO2/0.75 bar N2 mixture, mmen-CuBTTri adsorbs 2.38 mmol CO2 g−1 (9.5 wt%) with a selectivity of 327, as determined using Ideal Adsorbed Solution Theory (IAST). The high capacity and selectivity are consequences of the exceptionally large isosteric heat of CO2 adsorption, calculated to be −96 kJ mol−1 at zero coverage. Infrared spectra support chemisorption between amines and CO2 as one of the primary mechanisms of uptake. Despite the large initial heat of adsorption, the CO2 uptake was fully reversible and the framework could be easily regenerated at 60 °C, enabling a cycling time of just 27 min with no loss of capacity over the course of 72 adsorption/desorption cycles.
Introduction
The separation of carbon dioxide from nitrogen at low pressures, applicable to post-combustion carbon capture, has been extensively studied in porous solids.1 The majority of solid surfaces preferentially adsorb CO2 over N2via a physisorption mechanism, owing to the greater polarizability and quadrupole moment of CO2.2 However, the introduction of polarizing functional groups onto surfaces can enhance this selective adsorption. Accordingly, activated carbons, zeolites and metal–organic frameworks, all of which can be replete with strongly polarizing sites, are among the best solid adsorbents for CO2 separations. In particular, metal–organic frameworks show exceptional promise for such applications, due to their high surface areas and chemical tunability.3For post-combustion CO2 capture, maximizing adsorption capacity for CO2 at low pressures is highly desirable. Because the partial pressure of CO2 in flue gas emitted from coal-fired power stations is typically between 0.10 and 0.15 bar,4 the simplest approximation for the capacity of materials being considered is the quantity of gas adsorbed at these lower pressures, not the capacity at 1 bar. To date, the best performing materials in this regard have incorporated coordinatively-unsaturated metal centers acting as Lewis acids.5,6 Most notably, five-coordinate Mg2+ cations in Mg2(dobdc) (dobdc4− = 1,4-dioxido-2,5-benzenedicarboxylate) impart the framework with impressive characteristics for low-pressure CO2 adsorption: approximately 6.1 mmol g−1 at 0.15 bar CO2 pressure and 25 °C.6
Such solid adsorbents are being investigated as alternatives to the aqueous amine scrubbers traditionally used to effect CO2 removal from a mixed gas stream. In the simplest configuration of an amine scrubber, a gas mixture containing CO2 is passed through an aqueous solution of monoethanolamine (MEA).7 The formation of ammonium carbamate from two MEA molecules and one CO2 molecule endows the scrubber with extremely high selectivity for CO2, but significant energy is required to regenerate the solution. This high regeneration energy cost has two primary components: first, the strong, chemisorptive bond between the carbon dioxide and the amine must be broken; second, a large amount of spectator water solvent must be heated and cooled along with the active amine adsorbent, giving rise to an inefficient system.8 Because amines are corrosive to plant infrastructure, solutions are typically limited to no more than 30% (w/w) of the amine, and a significant increase in this concentration is not deemed feasible. In addition to the solvent boil-off that occurs during repeated regeneration cycles, these limitations represent the most significant obstacles to wider implementation of amine scrubbing technologies for post-combustion carbon capture. The National Energy Technology Laboratory, a unit of the United States Department of Energy, has established targets for CO2 capture materials. Implementation of carbon capture and storage utilizing amine scrubbers is expected to result in an 86% increase in the cost of electricity,8b while the current target is for a maximum 35% cost increase with 90% CO2 removal. For these reasons, new materials for post-combustion carbon capture are actively being sought.
Among the primary benefits of physisorption onto solid materials is the low regeneration energy compared to that required for aqueous amines; however, this benefit frequently comes at the expense of low capacity and poor selectivity. Our research therefore focuses on developing adsorbents that bridge the two approaches through the incorporation of sites that bind CO2via chemisorption into solid materials. By eliminating the aqueous solvent, we aim to develop new materials that have significantly lower regeneration costs compared with traditional amine scrubbers, yet maintain their exceptional selectivity and high capacity for CO2 at low pressures.
We previously reported the grafting of ethylenediamine (en) within the water-stable metal–organic framework H3[(Cu4Cl)3(BTTri)8] (CuBTTri; H3BTTri = 1,3,5-tri(1H-1,2,3-triazol-4-yl)benzene).9 In this system, en-CuBTTri, the diamine was incorporated as a ligand on the Cu2+ cation sites exposed on the framework pore surfaces.10 Because the diamines were shorter than the distance between two adjacent metal sites, it was proposed that one amine from each en molecule was bound to a single metal site, while the other amine was free to interact with guest gas molecules upon framework activation. At zero coverage, the isosteric heat of CO2 adsorption approached −90 kJ mol−1, significantly greater in magnitude than the value of −20 kJ mol−1 originally calculated for the unappended framework. The large isosteric heat was attributed to the formation of a weak bond between an amine and a CO2 molecule, most likely forming a zwitterionic carbamate. Unfortunately, only a very low CO2 capacity could be achieved in this material, presumably owing to clogging of the outermost framework pores within each crystallite upon en grafting.
The incorporation of alkylamine groups at higher loadings is expected to further polarize the overall surface area of a metal–organic framework, thereby increasing the capacity for CO2 capture. Other functional groups are similarly capable of polarizing framework surfaces,11 but few are capable of undergoing the chemisorptive type process we are pursuing. Inspired by the current generation of amine scrubbers that have begun to incorporate sterically hindered amines for improved performance, we were interested in surveying the influence of amine sterics on the isosteric heat of adsorption.12 Indeed, higher-order amines, in particular secondary amines, have been reported to have more favorable adsorption characteristics in solutions as well as on solid adsorbents.13 Herein, we demonstrate that the incorporation of N,N′-dimethylethylenediamine (mmen) at high loadings within CuBTTri affords a material with exceptional CO2 capture characteristics.
Results and discussion
Synthesis of mmen-CuBTTri
In contrast to en, conditions for achieving a high loading of mmen in CuBTTri were readily identified. A sample of the metal–organic framework was suspended in 10 mL of anhydrous hexane, and upon addition of 1.8 equiv. of mmen, the color of the solid instantaneously changed to a deep blue. To effect complete grafting of the diamine, the suspension was refluxed for 18 h under an N2 atmosphere. The resulting purple solid was collected by filtration and washed copiously with hexanes. The grafted material, mmen-CuBTTri, was activated by heating at 50 °C for 24 h under a dynamic vacuum prior to gas adsorption. Nitrogen adsorption isotherms collected at 77 K indicate a BET surface area of 870 m2 g−1, while powder X-ray diffraction data show the structure of the CuBTTri framework to be intact. Overall, the characterization data are most consistent with a chemical formula of H3[(Cu4Cl)3(BTTri)8(mmen)12], with approximately one mmen molecule for each available metal site. Thus, mmen-CuBTTri is thought to possess a high concentration of surface-appended mmen molecules, where one of the amine groups is bound to a Cu2+ center, while the other dangles within the pore, as depicted in Fig. 1.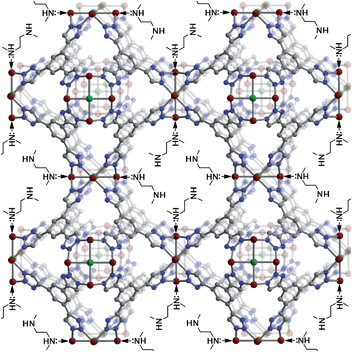 | ||
| Fig. 1 A portion of the structure of the amine functionalized metal–organic framework mmen-CuBTTri, with red, green, blue and gray spheres representing Cu, Cl, N and C atoms, respectively. Stoichiometric incorporation of the diamineN,N′-dimethylethylenediamine onto open metal sites within the pores begets a framework with excellent capacity and selectivity for CO2 capture at low pressures. | ||
CO2 and N2 uptake
The incorporation of mmen in CuBTTri results in a material with excellent CO2 adsorption characteristics. As shown in Fig. 2, mmen-CuBTTri displays significantly enhanced CO2 adsorption at all pressures between 0 and 1.1 bar relative to the unappended framework. The previously reported material en-CuBTTri, with a loading of just 0.3 en per Cu2+ center, showed increased CO2 adsorption only at pressures less than 0.08 bar.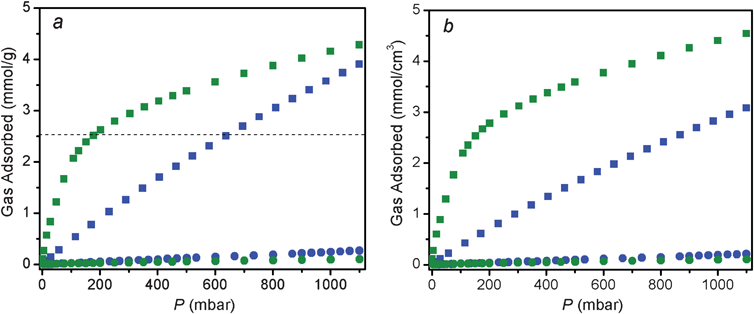 | ||
| Fig. 2 Isotherms for CO2 (squares) and N2 (circles) adsorption at 25 °C for mmen-CuBTTri (green) and CuBTTri (blue). Gravimetric capacity (a, left) and volumetric capacity (b, right) are plotted together for comparison. The horizontal dashed line in (a) corresponds to 10 wt% CO2 adsorption. | ||
Quantifying the extent of the improvement in CO2 adsorption between CuBTTri and mmen-CuBTTri is not trivial, since the degree of improvement depends significantly on the units to which the gas uptake is normalized. Fig. 2a plots the gravimetric gas adsorption isotherm, while Fig. 2b plots the crystallite volumetric gas adsorption isotherm for the two materials. Gravimetric capacities were converted into volumetric capacities via unit cell densities. Experimentally, gravimetric capacity is significantly easier to measure than volumetric capacity and gas sorption data normalized to mass is widely reported. The volumetric capacity for an actual adsorber unit is dependent upon how crystallites pack together and the fraction of void space within the occupied volume. Yet, gravimetric capacity alone does not provide a complete measure of the performance of a material being proposed for stationary applications, such as post-combustion CO2 capture. Here, infrastructure costs are linked more directly to the volume the adsorbent would occupy than to its mass. Because incorporation of mmen into CuBTTri increases the framework density by 34% with no significant change in volume, this system is a good candidate for comparisons between gravimetric and volumetric capacities. Density calculations for CuBTTri and mmen-CuBTTri are presented in the ESI†. It is important to note, however, that no single-crystal diffraction data are available for either CuBTTri or mmen-CuBTTri. Framework volumes are based upon powder pattern unit cell optimizations, and framework compositions are based upon elemental and thermogravimetric analyses.
At 25 °C and 1 bar, mmen-CuBTTri adsorbs 4.2 mmol g−1 of CO2 (15.4 wt%), representing a 15% improvement in gravimetric capacity compared to the unmodified CuBTTri framework. The best metal–organic framework for storing CO2 at 1 bar, Mg2(dobdc), adsorbs ca. 8.5 mmol g−1 at 25 °C.6 However, CO2 comprises at most 15% of coal fired power station flue gas and the effluent is released into the environment at total pressures near 1 bar. Thus, the more important criterion for CO2 capacity is that of the framework at a pressure near 0.15 bar. At 25 °C and 0.15 bar of CO2, mmen-CuBTTri adsorbs 2.38 mmol g−1 (9.5 wt%). Note that 2.90 mmol g−1 would correspond to the adsorption of one CO2 molecule per mmen in the functionalized framework. Under the same conditions, the unmodified framework only adsorbs 0.69 mmol g−1 (2.9 wt%). Thus, on a gravimetric basis, mmen-CuBTTri adsorbs nearly 3.5 times as much CO2 at the relevant pressures. Volumetrically, however, mmen-CuBTTri adsorbs about 4.7 times more CO2 at 0.15 bar than CuBTTri. The difference between gravimetric and volumetric capacities is a direct consequence of the increased mass of the appended framework over the unappended material.
At 25 °C, mmen-CuBTTri adsorbs less N2 than CuBTTri at all pressures between 0 and 1.1 bar. This is due to the reduction in specific surface area upon incorporation of mmen, with the BET surface area of 870 m2 g−1 for mmen-CuBTTri being roughly half of the 1770 m2 g−1 observed for CuBTTri.9 The additional polarizing sites in mmen-CuBTTri enhance N2 adsorption less than the decreased surface area diminishes N2 adsorption. The opposite trend was observed for CO2 adsorption. Enhanced adsorption of only one gas is a defining characteristic of chemisorption.14 In contrast, frameworks replete with open metal cation sites can be expected to polarize all gases more effectively, including N2, accounting for the substantially greater N2 adsorption in Mg2(dobdc) relative to mmen-CuBTTri.
The selectivity (S) for adsorption of CO2 over N2 in mmen-CuBTTri was calculated from the single-component isotherm data. For CO2 capture, this value typically reports the ratio of the adsorbed amount of CO2 at 0.15 bar to the adsorbed amount of N2 at 0.75 bar; the value is normalized for the pressures chosen, according to eqn (1).
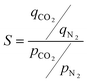 | (1) |
The values are derived from an approximate flue gas composition of 15% CO2, 75% N2 and 10% other gases, at a total pressure of 1 bar. Pure-component isotherm selectivities, which frequently are calculated from the excess adsorption data directly measured by gas adsorption, can be misleading. The adsorption selectivity, SIAST, was therefore modeled by applying the Ideal Adsorbed Solution Theory (IAST) to the calculated absolute adsorption isotherms.15 The accuracy of the IAST procedure has been established for the adsorption of a wide variety of gas mixtures in different zeolites, as well as CO2 capture in metal–organic frameworks.16 For mmen-CuBTTri, SIAST values for a mixture of 0.15 bar CO2 and 0.75 bar N2 at three temperatures were calculated to be 327 (25 °C), 200 (35 °C), and 123 (45 °C). To the best of our knowledge, the selectivity observed at 25 °C is the highest value yet reported for a metal–organic framework.
Isosteric heat of adsorption and working capacity
Utilizing a dual-site Langmuir adsorption model, isosteric heats of adsorption were calculated for CO2 in mmen-CuBTTri. Details of the model can be found in the ESI†. Fig. 3 compares these values to those obtained from data for bare CuBTTri, which were fit using a single-site Langmuir model to give a value of −24 kJ mol−1. The N2 adsorption isotherm for mmen-CuBTTri was also fit to a single-site Langmuir model, resulting in a calculated isosteric heat of adsorption of −15 kJ mol−1.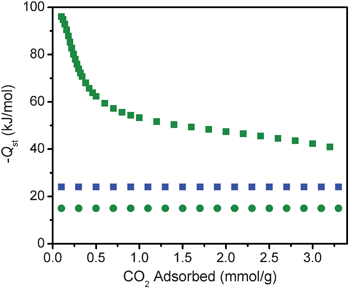 | ||
| Fig. 3 Isosteric heats of adsorption for CO2 (squares) and N2 (circles) in mmen-CuBTTri (green) and CuBTTri (blue), as obtained from fits to the gas adsorption data collected at 25, 35 and 45 °C. | ||
The isosteric heat of CO2 adsorption in mmen-CuBTTri approaches −96 kJ mol−1 at zero coverage, corresponding to the largest value yet reported for CO2 adsorption in a metal–organic framework. We previously reported a value of −90 kJ mol−1 for CO2 adsorption in en-CuBTTri, as calculated by fitting the Clausius–Clapeyron relation to excess adsorption values. For comparison to mmen-CuBTTri, the heat of adsorption for en-CuBTTri was recalculated with the same dual-site Langmuir model. Because this model incorporates absolute adsorption, direct comparisons between the two different models are not possible. From the dual-site Langmuir model, we calculate the isosteric heat of CO2 adsorption in en-CuBTTri to be −78 kJ mol−1 at zero coverage (see Fig. S9, ESI†), nearly 20 kJ mol−1 lower in magnitude than the heat calculated for mmen-CuBTTri.
We do not attribute the greater heat of adsorption of mmen-CuBTTri to any enhanced adsorption characteristics of secondary amines. Rather, mmen-CuBTTri has a significantly larger number of free amines available to bind guest CO2 molecules. Because isosteric heats correspond to the average of all adsorption sites potentially populated at a specific coverage level, at zero coverage there is a higher probability of the CO2 molecule adsorbing onto an amine in mmen-CuBTTri compared with en-CuBTTri. Despite the large binding enthalpy between alkylamines and CO2, the substantial decrease in the −Qst data with loading indicates that many weaker adsorption sites are also being sampled by the CO2 molecules under the conditions probed. While amine sterics may play a role in the improved adsorption properties of mmen-CuBTTri, we cannot yet make comparisons between isosteric heats of adsorption as a function of amine sterics because of the differences in amine loading levels.
Isosteric heats of adsorption are commonly calculated after fitting adsorption equilibrium data to the Virial equation. Fig. S8, ESI†, overlays the isosteric heats of adsorption calculated for mmen-CuBTTri using both the dual-site Langmuir and the virial methods. At zero coverage, the virial method gives a significantly lower magnitude for the heat of adsorption: −66 kJ mol−1. As shown in Fig. S10, ESI†, however, inflections in the isotherms for mmen-CuBTTri are not accurately modeled with the virial method. We therefore believe the values of the dual-site model are significantly more accurate at low CO2 loadings.17 In contrast, at intermediate loadings, there is good agreement between the dual-site Langmuir and virial fits. While mmen-CuBTTri exhibits a large heat of adsorption at zero loading, heats of adsorption at intermediate loadings are of course also important for CO2 capture. At a loading of 2.4 mmol g−1, the approximate capacity of mmen-CuBTTri for CO2 at 0.15 bar, the isosteric heat of adsorption was calculated to be about −45 kJ mol−1 by both the dual-site Langmuir and virial models. Hence, in CO2 capture applications, the average enthalpy of adsorption for CO2 would be significantly less than the −96 kJ mol−1 value calculated for very low coverage levels. This has important implications for adsorbent regeneration.
Solid adsorbents with large isosteric heats of adsorption have considerable advantages including high selectivity and high capacity for CO2 at low partial pressures; however, they are often believed to be the most difficult to regenerate. Regeneration, however, is very dependent on the method best suited to a given material. For carbon capture from flue gas streams, vacuum and temperature swing adsorption methodologies are the ones most frequently envisioned. Vacuum swing adsorption can best be approximated by the difference in capacities between the adsorption and desorption pressures. For mmen-CuBTTri, we calculate a ca. 7 wt% working capacity between 0.15 and 0.02 bar at 25 °C. For temperature swing methods, adsorbents with high heats of adsorption may prove to be better candidates than materials with moderate heats of adsorption. This is because the capacities of adsorbents with high heats of adsorption are more dependent on temperature than materials with smaller heats of adsorption.18 The significantly greater working capacities of strongly binding adsorbents may lead to materials that are less expensive to operate than those that have smaller working capacities.
We sought to test the cyclability of the material as a CO2 adsorbent using a combined temperature swing and nitrogen purge approach. Utilizing a thermogravimetric analyzer, a mixture of 15% CO2 in N2 was introduced into the furnace for 5 min at 25 °C. As shown in Fig. 4, the sample mass increased by nearly 7% upon introduction of the mixed gas, due to strong adsorption of CO2, even in the dilute mixture. The adsorbent was then regenerated by changing the flow to a pure N2 stream followed by rapid ramping of the furnace at 5 °C min−1 to 60 °C. The sample was held for 2 min at 60 °C, and then cooled at 5 °C min−1 to 25 °C. The temperature was allowed to stabilize for 2 min, followed by the reintroduction of the 15% CO2 in N2 mixture. The 27 min cycling procedure was repeated 72 times, with no apparent change in capacity. The kinetics of the adsorption are sufficiently quick that little additional CO2 is adsorbed after the first few minutes, and even shorter cycle times could be utilized with only small reductions in capacity. Similarly, complete desorption is realized prior to the end of each 2 min isotherm. Importantly, the cycling capacity of mmen-CuBTTri under dry conditions is comparable to or even greater than the working capacity of a 30% MEA solution, which is frequently reported as 5.5 wt%.19
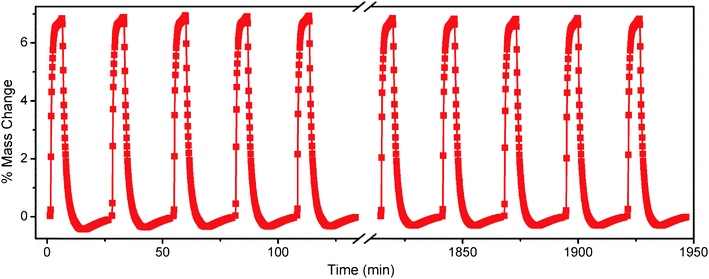 | ||
| Fig. 4 Upon introduction of a 15% CO2 mixture in N2 at 25 °C, the mass of mmen-CuBTTri increased by nearly 7% as measured by thermogravimetric analysis. Upon saturation, a N2 purge flow with a temperature swing to 60 °C fully regenerated the material with no apparent capacity loss after 72 cycles. Note that the data are uncorrected for changes in buoyancy, which are small compared to changes in sample mass; this accounts for the effective negative mass of the sample at 60 °C. The sample mass was normalized to 0% at 25 °C under a flowing N2 atmosphere. | ||
Infrared spectra
Heats of adsorption approaching −100 kJ mol−1 and highly specific interactions are indicative of chemisorptive processes. In the absence of water, we believe that the electrophilic CO2 molecule is accepting electron density from the lone pair of the free amine of mmen, forming a zwitterionic carbamate. Previous spectroscopic studies of CO2 binding in amine-containing metal–organic frameworks have investigated only less basic aromatic amines.20 For example, it was recently shown in NH2-MIL-53(Al) that the amine was not directly interacting with the CO2, but rather, other physisorptive processes account for the favorable adsorption characteristics of the material.21Diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) measurements were performed on mmen-CuBTTri using a high-pressure (0–3 bar) gas cell to confirm and characterize the proposed chemisorptive process. Fig. 5 plots the infrared absorbance of mmen-CuBTTri under various pressures of CO2. Most notably, a total disappearance of the N–H band at 3283 cm−1 is clearly observed upon introduction of 5% CO2 in He into the cell at increasing pressures. The reported frequency for the N–H stretch in free mmen is 3279 cm−1.22 Upon regeneration of the solid under vacuum and heating, the N–H stretching band returns. A sharp band at 1386 cm−1 also appears in the spectra as CO2 is introduced; diffuse bands near 1669, 1487 and 1057 cm−1 are also apparent in addition to changes in the fingerprint region.
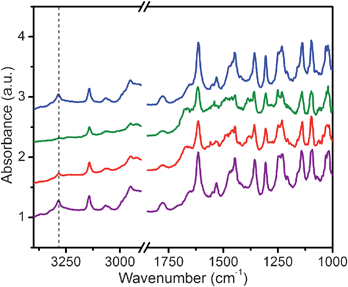 | ||
| Fig. 5 Infrared spectra obtained upon exposure of mmen-CuBTTri to a 5% CO2/95% He gas mixture in a high-pressure DRIFTS cell. Under dry conditions, the N–H stretch of mmen is apparent at 3283 cm−1 (vertical dashed line) on a fully evacuated sample (purple). Dilute CO2 in He was slowly introduced into the cell (red) up to a dynamic pressure of 1.5 bar (green). Upon saturation, the N–H stretch fully disappeared. Following reactivation under vacuum and heating at 60 °C (blue), the N–H stretch reappeared. | ||
Water was strictly excluded from the material for the DRIFTS measurements to eliminate the possibility of carbonate formation. Because the amines are tethered to the framework and well separated, we do not believe that it is possible for two amines to be concertedly interacting with a single CO2 molecule. Similar measurements have been performed on other porous materials, most notably amine-grafted mesoporous silicas.23 In these materials, however, two amines can often act concertedly and the experiments frequently incorporated water vapor, making direct comparisons difficult. It has been previously reported, however, in dry amino acid-based ionic liquids that zwitterionic CO2− species exhibit a sharp infrared band near 1660 cm−1.24 From the large calculated heat of adsorption and observed band changes in the infrared region upon CO2 addition, we believe that the primary mechanism of CO2 adsorption at low pressures is the chemisorption of CO2 gas onto mmen molecules, resulting in the formation of zwitterioinc carbamates or carbamic acid.
Steric effects on diamine incorporation
The framework CuBTTri was additionally modified with N-methylethylenediamine (men), an asymmetric diamine with one primary and one secondary amine. Relative to the performance of mmen-CuBTTri, men-CuBTTri performs more similarly to en-CuBTTri, for which only a small enhancement in CO2 uptake was observed at very low pressures. A small improvement in performance was, however, realized through the use of men over en. Additional information about the synthesis, characterization, and gas sorption properties of men-CuBTTri has been included in the ESI†.The significantly greater adsorption of CO2 in mmen-CuBTTri is attributable to the larger number of amines that are accessible to guest CO2 molecules. There are two primary factors affecting the incorporation of primary amines into CuBTTri. First, the best fits to the elemental analysis data indicate at least two times as many diamine molecules were incorporated into mmen-CuBTTri vs. the en- and men-analogues. A reasonable explanation for the higher incorporation of mmen into the framework is the formation of a weaker coordinate bond between the secondary amine on mmen and a framework Cu2+ ion compared with the relatively stronger coordinate bond formed between a primary amine and a Cu2+ ion. The significantly greater reversibility of mmen binding with Cu2+ imparts it with a faster diffusion rate through the pores such that mmen molecules that bonded with copper sites near pore openings were labile in the hot hexane solution employed for grafting. These diamines were capable of migrating deeper into pores to achieve high surface coverage. In the case of primary amines, the irreversibility of the coordinate bond at the synthesis conditions surveyed severely limited the diffusion of en and men into the pores. Once appended, additional diamines could only diffuse through constricted pores to reach interior metal sites. Note that in men-CuBTTri, the primary amine end of men is coordinated to the metal centers while the sterically hindered secondary amine is available to interact with guest molecules in the pores. Increased reversibility of en and men binding was sought through the use of higher boiling solvents and lower concentrations; however, grafting temperatures that significantly exceeded 100 °C resulted in the decomposition of the framework.
Second, based upon the calculated heats of adsorption for en- and men-CuBTTri (see Fig. S9, ESI†), the number of amines that were strongly adsorbing CO2 was significantly less than the number of amines that were appended in each framework. This is because not all of the amines in the en- and men- frameworks bonded to open metal sites. Grafted amines severely reduced the pore diameters, slowing diffusion rates as previously discussed. Some of these pores then became blocked by the excess amines. Amines and other adsorption sites beyond these blockages were inaccessible to guest gases. This was confirmed by the low surface areas calculated for en- and men-CuBTTri (see Table S6, ESI†). By comparison, the surface area for mmen-CuBTTri was significantly higher despite the greater number of total amines calculated to be within the framework.
Future work will focus on confirming the proposed mechanism for amine migration through the pores as well as identifying other metal–organic frameworks that are more suitable for incorporation of diamines containing primary amines.
Conclusions
The incorporation of alkylamine functional groups onto surfaces can greatly improve the CO2 adsorption characteristics of metal–organic frameworks. Amines do not necessarily simply polarize CO2; rather, they strongly and selectively bind it via chemisorptive interactions. The new compound mmen-CuBTTri has the highest selectivity and binding strength for CO2 of any metal–organic framework reported to date. Despite its large isosteric heat of CO2 adsorption, the material can quickly be regenerated using mild temperature swings.Amines tethered to solid surfaces within porous materials have considerable advantages over aqueous alkanolamines. Understanding the effects of other gases on CO2 adsorption, especially water vapor, is important for all metal–organic frameworks being considered for CO2 capture, and studies to that end for mmen-CuBTTri are currently underway.
Acknowledgements
This research was funded through the Center for Gas Separations Relevant to Clean Energy Technologies, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award No. DE-SC0001015, and was further supported in its initial stages by the Sustainable Products & Solutions Program at the University of California, Berkeley.Notes and references
- (a) S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS; (b) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef CAS.
- (a) K. B. Lee, M. G. Beaver, H. S. Caram and S. Sircar, Ind. Eng. Chem. Res., 2008, 47, 8048 CrossRef CAS; (b) P. Chowdhury, C. Bikkina and S. Gumma, J. Phys. Chem. C, 2009, 113, 6616 CrossRef CAS , and references therein.
- (a) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (b) S. Keskin, T. van Heest and D. Sholl, ChemSusChem, 2010, 3, 879 CrossRef CAS.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9 CrossRef CAS.
- (a) P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. D. Weireld, J.-S. Chang, D.-Y. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245 CrossRef; (b) A.Ö. Yazaydin, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425 CrossRef; (c) A. Yazaydın, R. Q. Snurr, T.-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 18198 CrossRef CAS; (d) D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637 CrossRef CAS; (e) E. D. Bloch, D. Britt, D. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382 CrossRef CAS; (f) K. Sumida, S. Horike, S. S. Kaye, Z. R. Herm, W. L. Queen, C. M. Brown, F. Grandjean, G. J. Long, A. Dailly and J. R. Long, Chem. Sci., 2010, 1, 184 RSC; (g) A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368 CrossRef CAS.
- (a) S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870 CrossRef CAS; (b) P. D. C. Dietzel, V. Besikiotis and Richard Blom, J. Mater. Chem., 2009, 19, 7362 RSC; (c) J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011 10.1039/C1EE01720A.
- G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS.
- (a) T. Fout and J. T. Murphy, DOE/NETL's Carbon Capture R&D Program for Existing Coal-Fired Power Plants. Report DOE/NETL-2009/1356, U.S. DOE National Energy Technology Laboratory, Pittsburgh, February 2009 Search PubMed; (b) J. Ciferno, M. Matuszewski, J. J. Marano and S. Chen, Existing Plants, Emissions and Capture–Setting CO2 Program Goals. Report DOE/NETL-2009/1366, U.S. DOE National Energy Technology Laboratory, Pittsburgh, April 2009 Search PubMed; (c) J. Ciferno, J. Litynski, S. Plasynski, J. Murphy, G. Vaux, R. Munson and J. Marano, DOE/NETL Carbon Dioxide Capture and Storage RD&D Roadmap, U.S. DOE National Energy Technology Laboratory, Pittsburgh, December, 2010 Search PubMed.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, V. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS.
- (a) Y.-S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131 RSC; (b) A. Torrisi, R. G. Bell and C. Mellot-Draznieks, Cryst. Growth Des., 2010, 10, 2839 CrossRef CAS; (c) R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173 RSC.
- (a) K. S. Fisher, G. T. Rochelle and C. Schubert, Advanced Amine Solvent Formulations and Process Integration for Near-Term CO2 Capture Success. Final Report to U.S. DOE, DE-FG02–06ER84625. Trimeric Corporation, Texas, 2007 Search PubMed; (b) R. Hook, Ind. Eng. Chem. Res., 1997, 36, 1779–1790 CrossRef CAS.
- (a) 2003 ; (b) 2007 ; (c) M. L. Gray, Y. Soong, K. J. Champagne, H. Pennline, J. P. Baltrus, R. W. Stevens Jr., R. Khatri, S. S. C. Chuang and T. Filburn, Fuel Process. Technol., 2005, 86, 1449 CrossRef CAS; (d) V. Zelenak, D. Halamova, L. Gaberova, E. Bloch and P. Llewellyn, Microporous Mesoporous Mater., 2008, 116, 358 CrossRef CAS.
- R. L. Burwell, Pure Appl. Chem., 1975, 46, 71.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121 CrossRef CAS.
- (a) R. Krishna, S. Calero and B. Smit, Chem. Eng. J., 2002, 88, 81 CrossRef CAS; (b) R. Krishna and J. M. van Baten, Chem. Eng. J., 2007, 133, 121 CrossRef CAS; (c) R. Krishna and J. M. van Baten, Phys. Chem. Chem. Phys., 2011, 13, 10593 RSC.
- T. J. H. Vlugt, R. Krishna and B. Smit, J. Phys. Chem. B, 1999, 103, 1102 CrossRef CAS.
- A. H. Berger and A. S. Bhown, Energy Proc., 2011, 4, 562 Search PubMed.
- A. N. M. Peeters, A. P. C. Faaij and W. C. Turkenburg, Int. J. Greenhouse Gas Control, 2007, 1, 396 CrossRef CAS.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650 CrossRef CAS.
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Langmuir, 2011, 27, 3970 CrossRef CAS.
- E. Pretsch, P. Buhlmann and M. Badertscher, Structure Determination of Organic Compounds, 4th edn, Springer, Berlin, 2009 Search PubMed.
- (a) R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Ind. Eng. Chem. Res., 2005, 44, 3702 CrossRef CAS; (b) N. Hiyoshi, K. Yogo and T. Yashima, Microporous Mesoporous Mater., 2005, 84, 357 CrossRef CAS.
- J. Zhang, S. Zhang, K. Dong, Y. Zhang, Y. Shen and X. Lv, Chem.–Eur. J., 2006, 12, 4021 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, activation, calculations, fitting parameters, gas adsorption data, and additional figures. See DOI: 10.1039/c1sc00354b |
| This journal is © The Royal Society of Chemistry 2011 |