DOI:
10.1039/C1SC00405K
(Edge Article)
Chem. Sci., 2011,
2, 2367-2372
Silene equivalents through the rhodium-catalysed reactions of α-hypersilyl diazoesters: a computational and experimental study†
Received
28th June 2011
, Accepted 7th September 2011
First published on 22nd September 2011
Abstract
The generation of silenes through the rhodium-catalysed decomposition of α-hypersilyl diazocarbonyl compounds has been explored both computationally and experimentally. This transformation proceeds via a pathway involving initial formation of the carbene, followed by rearrangement, initially to a silene and ultimately to a ketene. Density functional theory (DFT) calculations of model compounds suggested that silene formation was most preferential with electron donating substituents attached to the carbonyl group. The predictions were experimentally evaluated and hypersilyl diazoacetates provided an unusually long-lived species (t1/2 > 40 h) that reacts as a formal silene equivalent. Further DFT calculations support the formation of an internally stabilised silene in the form of a 1,2-silaoxetene. Importantly the acylsilene–silaoxetene reaction is reversible and consequently this silene equivalent reacts with α,β-unsaturated carbonyl compounds to form cyclic silyl enol ethers which have considerable potential for further synthetic transformations.
Introduction
Reflecting their unique reactivity profile, silenes, compounds containing a silicon–carbon double bond, have been the subject of numerous studies.1 Much of this effort has been focused on fundamental aspects of structure and bonding with a particular emphasis on the preparation of stable isolable silenes. In most cases silenes are highly reactive, having a transient existence, and are commonly characterised through the adducts from reactions with a large variety of functionalities, including alcohols, alkenes and carbonyl compounds. Despite the broad repertoire of chemistry displayed by silenes there has been little effort to exploit the unique reactivity of these species in organic synthesis.2 In efforts to address this issue we have been exploring the application of transient silenes to the preparation of highly functionalised organosilanes and their subsequent elaboration to lactones, tetralols, cyclopropanes, etc.3 A major challenge to this goal has been the fact that although many methods for silene generation have been described, these tend not to be suitable for general preparative purposes, as they require reagents or conditions that are incompatible with many functional groups. In this paper we describe a potential solution to this challenge through the generation of a surprisingly stable silene equivalent, via the metal catalysed decomposition of α-hypersilyl diazo esters, and their subsequent reaction with α,β-unsaturated carbonyl compounds to afford substituted cyclic silylenols under very mild conditions.
Results and discussion
Synthetic approaches to silenes date to 1912 when Schlenk and Renning first claimed an approach through the dehydration of diphenylsilylmethanol.4 This report was subsequently shown to be erroneous and the first synthesis was not realised until 1967, when Gusel'nikov and Flowers unambiguously demonstrated the transient existence of silenes in the pyrolysis of dimethylsilacyclobutane.5 Since then many other methods have been described including the photochemical or thermal decomposition of α-silyl diazoalkanes.6 Similar transformations are possible from α-silyldiazocarbonyl compounds 1 although in these cases formation of silene 2 competes with the Wolff rearrangement to give ketene 3 (Fig. 1).7 Moreover, the silene is only transiently stable under these reaction conditions, rapidly rearranging to give a variety of products, including the alternative ketene 4, depending upon the nature of both the precursor structure and conditions used. Reflecting this observation no attempt has been described to use this transformation in a wider synthetic context.
 |
| | Fig. 1 Competing pathways for photolysis and thermolysis of α-silyldiazocarbonyl compounds 1. | |
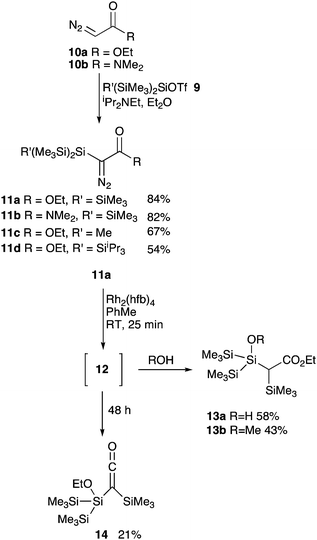
Mindful of these precedents, we were curious as to whether careful selection of the silene substituents coupled with milder conditions for silene generation, exploiting recent developments in metallocarbenoid chemistry,8 might enable silenes to be generated more effectively. In particular, the presence of a polysilyl-substituted silicon centre was predicted to be beneficial for the stability of the silene.2b,9 Prior to undertaking preparative work, a DFT study analysing the relative energies of the three possible products of an initially generated model carbene 6 for different carbonyl derivatives was undertaken (Fig. 2).10–12 Although this analysis ignores the activation energies for the competing pathways, these studies clearly revealed that the ester and amide derived silicon substituted silenes 7a and 7b have lower relative energies, when compared to the Wolff rearrangement product 5a and 5b. In addition, given the known propensity for α-silyl-α-keto carbenes and carbenoids to undergo rapid conversion to ketenes 5,7a we opted to focus our efforts on the metal-catalysed chemistry of the amide and ester variants 11a and 11b (Scheme 1). However, since these silenes that result still have the potential to isomerise to the alternative, and thermodynamically more stable, ketenes 8, it was not known whether they could be efficiently trapped. The preparation of these followed standard protocols involving reaction of the parent α-diazocarbonyl compounds 10a and 10b with hypersilyltriflates 9,13 generated, in turn, from the reaction of the respective phenyl silanes14 with triflic acid. With these precursors in hand, attention turned to the generation of the carbene exploring a range of metal complexes derived from palladium, copper and rhodium salts.15
 |
| | Fig. 2 Calculated relative energies (B3LYP/6-311+G(d,p)) of possible products generated from a model α-hypersilylcarbene 6 in its singlet state. | |
 |
| | Scheme 1 Synthesis and reactivity of α-silyldiazocarbonyl compounds. | |
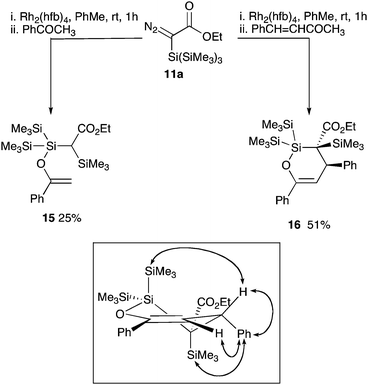
Whilst complexes derived from the first two of these metals proved ineffective at promoting clean conversion to the carbene or subsequent products, treatment of the diazoester 11a with the electrophilic rhodium complex, Rh2(hfb)4 (hfb = heptafluorobutyrate), led to a very rapid conversion to a new species 12 as evidenced by the appearance of signals at 35.6, −15.3 and −17.1 ppm in the 29Si NMR spectrum (C6D6, −80 °C, 140 MHz). Significantly, treatment of this solution with nucleophiles leads to the formation of the silanes (13) that would be expected to arise from the reaction of a transient silene. In contrast to these observations, Maas has demonstrated that CuOTf efficiently converted trimethylsilyldiazoacetate to the carbene dimer, whilst Rh2(pfb)4 leads to the direct formation of the silene derived ketene.7g Presumably the former reflects the higher steric demands of the hypersilyl group and the latter the increased stability of the hypersilyl silene. In contrast to the preliminary DFT predictions, in which the silene derived from a diazoacetamide was predicted to be more energetically favourable, all attempts to activate 11b either returned unchanged starting materials or led to an intractable and complex product mixture. Surprisingly, given the very low steric bulk surrounding the silicon atom and the formal polarity of the proposed silicon carbon double bond, in solution the species 12 derived from diazo ester 11a appeared to be relatively stable showing only slow decomposition to the ketene 14 (δSi 6.6, 5.7 and −13.3 ppm; δC 165.6 ppm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)) dependent on the concentration (t1/2 = 80 h, c = 0.028 M; t1/2 = 43 h, c = 0.042 M in toluene). Curiously this half-life also seemed highly dependent of the polarity of the solvent, with much more rapid decomposition to the ketene occurring in more polar electron donating solvents (relative rate PhMe < 1,4-dioxane < THF < MeCN < Et3N < pyridine). This relatively prolonged lifetime permitted facile evaluation of this species as a reagent for further transformations (Scheme 2). Following rhodium-catalysed decomposition of diazo silyl ester 11a, addition of a selection of known silenophiles led to varied reaction outcomes. Addition of simple hydrocarbon based silenophiles such as phenyl acetylene, 2,3-dimethyl butadiene and styrene led only to the observation of silanol 13a in the crude reaction mixture with no evidence for any cycloadducts. In contrast, reaction with the polar silenophiles acetophenone and chalcone afforded the isolable silyl enol ethers 15 and 16, respectively (Scheme 2) as would be expected for the reaction of a transient silene. Interestingly, from a synthetic perspective, the cyclic silyl enol ether 16 was formed as a single diastereoisomer in which the Ph and SiMe3 groups were cis to each other as confirmed by NOESY experiments. Although the reactions with alcohols and carbonyl compounds is suggestive of the presence of a silene intermediate, the presence of only three discrete silicon signals in the 29Si NMR spectrum of the initial reaction mixture was not consistent with such a structure. Alternative silene derivatives, including the silaoxetene 12 and the “silene dimer” 21 (Fig. 3), which would accommodate this data have precedent in other studies, albeit not from ester containing starting materials.7h,i,16,17 Whilst these might be anticipated to undergo reversion to a silene-like structure on addition of a coordinating ligand (solvent/carbonyl group, etc.) attempts to detect or isolate these, either by mass spectrometry or low-temperature crystallisation techniques, were unsuccessful. In order to provide confirmation of the structure, calculations with the gauge including atomic orbitals (GIAO) method16 were undertaken to predict the 13C and 29Si NMR spectra of silene 17, dimer 18 and silaoxetene 12 (Fig. 3). Comparison of the experimental NMR data for compound 12 (C-3, δC = 67.0 ppm; C-4, δC = 161.1 ppm; Si-2, δSi = 35.6 ppm)) with that calculated for both silaoxetene 12 (C-3, δC = 67.9 ppm; C-4, δC = 170.5 ppm; Si-2, δSi = 46.3 ppm) and dimer clearly supports the suggestion that the former is the initial product of the rhodium-mediated diazo compound decomposition. To account for the outcome of the rearrangement of the carbene generated from diazo compounds 11a and 11b, the isomerisation of model carbenes 6a and 6b, was further investigated using DFT calculations (Fig. 4). The extrusion of N2 from α-silyl substituted diazo compounds leads to a silylcarbene 6, which readily undergoes transformation to the corresponding silene 7. Whilst, for computational simplicity, this analysis neglects the role of the metal in stabilising the initially formed carbene, the key aspect of the pathway is the fate of the silene, which is metal independent. This silene is short lived, rapidly forming silaoxetene 19. This appears to be the kinetically favourable product of this reaction and, given the relatively low calculated energy barriers, a slow reversion to the silene, and subsequently the formation of a thermodynamically stable silylketene 8 occurs over time. The enhanced rate for this interconversion in more polar solvents potentially relates to coordination or nucleophilic attack by the solvent at the electrophilic silicon atom of the silaoxetene, leading to an elongation of the Si–O bond, and hence a lower barrier to cycloreversion. However, there is relatively little difference between the profiles for amide 6b and ester 6a, and these calculations did not provide any obvious insights as to why extrusion of N2 from hypersilyl diazo amide 11b follows a different pathway to that of the ester 11a. Potentially, for the less electrophilic carbene generated from diazo compound 11b metal coordination may be more significant, lowering the rate of silene formation enabling alternative carbene/carbenoid processes to effectively compete.
O)) dependent on the concentration (t1/2 = 80 h, c = 0.028 M; t1/2 = 43 h, c = 0.042 M in toluene). Curiously this half-life also seemed highly dependent of the polarity of the solvent, with much more rapid decomposition to the ketene occurring in more polar electron donating solvents (relative rate PhMe < 1,4-dioxane < THF < MeCN < Et3N < pyridine). This relatively prolonged lifetime permitted facile evaluation of this species as a reagent for further transformations (Scheme 2). Following rhodium-catalysed decomposition of diazo silyl ester 11a, addition of a selection of known silenophiles led to varied reaction outcomes. Addition of simple hydrocarbon based silenophiles such as phenyl acetylene, 2,3-dimethyl butadiene and styrene led only to the observation of silanol 13a in the crude reaction mixture with no evidence for any cycloadducts. In contrast, reaction with the polar silenophiles acetophenone and chalcone afforded the isolable silyl enol ethers 15 and 16, respectively (Scheme 2) as would be expected for the reaction of a transient silene. Interestingly, from a synthetic perspective, the cyclic silyl enol ether 16 was formed as a single diastereoisomer in which the Ph and SiMe3 groups were cis to each other as confirmed by NOESY experiments. Although the reactions with alcohols and carbonyl compounds is suggestive of the presence of a silene intermediate, the presence of only three discrete silicon signals in the 29Si NMR spectrum of the initial reaction mixture was not consistent with such a structure. Alternative silene derivatives, including the silaoxetene 12 and the “silene dimer” 21 (Fig. 3), which would accommodate this data have precedent in other studies, albeit not from ester containing starting materials.7h,i,16,17 Whilst these might be anticipated to undergo reversion to a silene-like structure on addition of a coordinating ligand (solvent/carbonyl group, etc.) attempts to detect or isolate these, either by mass spectrometry or low-temperature crystallisation techniques, were unsuccessful. In order to provide confirmation of the structure, calculations with the gauge including atomic orbitals (GIAO) method16 were undertaken to predict the 13C and 29Si NMR spectra of silene 17, dimer 18 and silaoxetene 12 (Fig. 3). Comparison of the experimental NMR data for compound 12 (C-3, δC = 67.0 ppm; C-4, δC = 161.1 ppm; Si-2, δSi = 35.6 ppm)) with that calculated for both silaoxetene 12 (C-3, δC = 67.9 ppm; C-4, δC = 170.5 ppm; Si-2, δSi = 46.3 ppm) and dimer clearly supports the suggestion that the former is the initial product of the rhodium-mediated diazo compound decomposition. To account for the outcome of the rearrangement of the carbene generated from diazo compounds 11a and 11b, the isomerisation of model carbenes 6a and 6b, was further investigated using DFT calculations (Fig. 4). The extrusion of N2 from α-silyl substituted diazo compounds leads to a silylcarbene 6, which readily undergoes transformation to the corresponding silene 7. Whilst, for computational simplicity, this analysis neglects the role of the metal in stabilising the initially formed carbene, the key aspect of the pathway is the fate of the silene, which is metal independent. This silene is short lived, rapidly forming silaoxetene 19. This appears to be the kinetically favourable product of this reaction and, given the relatively low calculated energy barriers, a slow reversion to the silene, and subsequently the formation of a thermodynamically stable silylketene 8 occurs over time. The enhanced rate for this interconversion in more polar solvents potentially relates to coordination or nucleophilic attack by the solvent at the electrophilic silicon atom of the silaoxetene, leading to an elongation of the Si–O bond, and hence a lower barrier to cycloreversion. However, there is relatively little difference between the profiles for amide 6b and ester 6a, and these calculations did not provide any obvious insights as to why extrusion of N2 from hypersilyl diazo amide 11b follows a different pathway to that of the ester 11a. Potentially, for the less electrophilic carbene generated from diazo compound 11b metal coordination may be more significant, lowering the rate of silene formation enabling alternative carbene/carbenoid processes to effectively compete.
 |
| | Scheme 2
Silene-like reactivity of intermediate 12. Boxed figure shows selected NOESY correlations observed for 16. | |
 |
| | Fig. 3 Structures and calculated (GIAO-B3LYP/6-311+G(2d,p)//B3LYP/6-311G(d,p)) 13C and 29Si NMR chemical shifts for potential products of treatment of 11a with Rh2(hfb)4. | |
 |
| | Fig. 4 Calculated reaction pathway (B3LYP/6-311+G(d,p)) for the transformation of model carbene 6 to silaoxetene 22 and silylketene 8 showing relative electronic energies (kJ mol−1) for products, intermediates and predicted transition state structures. Values in parenthesis show corresponding ΔG298 energies. | |
Having established that rhodium-mediated decomposition of such α-hypersilyl diazoesters provides a simple and mild entry to silaoxetenes, that not only exhibit silene like reactivity but also have a relatively long lifetime, we then undertook a small survey of the scope of this transformation (Table 1). Using hypersilyl triflates 9c and 9d it was possible to modify the steric bulk around the silicon atom by the replacement of one of the trimethylsilyl groups with a methyl or triisopropylsilyl group. Whilst the former was readily reactive, the cycloadducts were formed as mixtures of diastereoisomers that proved challenging to purify. In the latter case, although no reaction could be observed in the presence of Rh2(pfb)4, presumably due to the increased steric hinderance, thermal or photochemical initiation led to the desired silaoxetene, albeit accompanied by competitive formation of a ketene end product of the reaction pathway. Interestingly for both substrates preferential migration of a trimethylsilyl group was observed. Given that this resulted in mixtures of diastereomeric products all further reactions were undertaken with the parent readily accessible tris(trimethylsilyl)silyl diazoester 11a. With this, a range of other α,β-unsaturated ketones were explored giving similar cyclic silyl enol ether adducts (Table 1). With the exception of the reaction with (E)-4-phenylbut-3-en-2-one in which a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of isomers was isolated, all products were formed with complete diastereoselectivity, as verified by 1H NMR analysis of the crude reaction mixture. A simple rationale for this selectivity is not obvious. On the basis that these products arise from a concerted, albeit highly asynchronous [4 + 2] cycloaddition, the formation of the observed product is consistent with a transition state in which the ester carbonyl group occupies an exo orientation to minimise unfavourable π–π interactions with the alkene of the silenophile. However, a stepwise ionic pathway involving initial coordination of the carbonyl group to the silene followed by “intramolecular” nucleophilic attack of a silicon substituted enolate to an activated enone is also plausible. In this case a model invoking steric interactions between the substituent on the α-carbon and the silene trimethyl silyl group is consistent with the observed outcome.
:1 mixture of isomers was isolated, all products were formed with complete diastereoselectivity, as verified by 1H NMR analysis of the crude reaction mixture. A simple rationale for this selectivity is not obvious. On the basis that these products arise from a concerted, albeit highly asynchronous [4 + 2] cycloaddition, the formation of the observed product is consistent with a transition state in which the ester carbonyl group occupies an exo orientation to minimise unfavourable π–π interactions with the alkene of the silenophile. However, a stepwise ionic pathway involving initial coordination of the carbonyl group to the silene followed by “intramolecular” nucleophilic attack of a silicon substituted enolate to an activated enone is also plausible. In this case a model invoking steric interactions between the substituent on the α-carbon and the silene trimethyl silyl group is consistent with the observed outcome.
Table 1 Cycloadducts arising from in situ reaction of siloxetene, generated from hypersilyldiazoacetates on treatment with Rh2(pfb)4 in toluene at rt, with α,β-unsaturated carbonyl compounds
|
a
Yield quoted for purified isolated products. bObtained as an unstable 2:1 mixture of diastereoisomers. cObtained following thermal reaction of 11d with chalcone. Unstable with respect to chromatography. |

|
With evidence to support the idea that a diazo decomposition pathway provides a mild route to a formal silene equivalent established, we then sought to demonstrate that the resulting cycloadducts could have synthetic utility. As a simple proof-of-concept study we examined the extrusion of the silicon centre from cycloadduct 16 (Scheme 3). Following precedents established in earlier studies, simple treatment of these cyclic enol ethers with a fluoride source led to rapid and efficient ring fragmentation. Use of triethylamine–trihydrofluoride complex resulted in complete desilylation to afford the 1,5-dicarbonyl compound 27. Presumably under these nucleophilic conditions rapid loss of the α-silicon atom occurs to generate a transient enolate. Attempts to trap this through an in situ aldol reaction with benzaldehyde proved unsuccessful. Alternatively, selective reaction of the siloxy silicon atom could be achieved through treatment with a combination of potassium hydrofluoride and trifluoroacetic acid at room temperature for a reduced reaction time of 17 h, leading to the formation of the α-silyl ester 28.
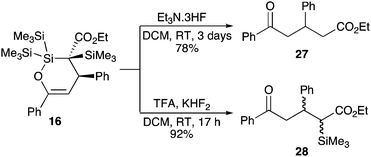 |
| | Scheme 3
Fluoride-mediated fragmentation of silacycle 16 to give 1,5-dicarbonyl compounds. | |
Conclusions
Silenes remain as reagents of considerable untapped potential for organic synthesis. A significant challenge in reaching this objective is the development of mild methods through which they can be generated and stored. The rhodium-catalysed decomposition of α-hypersilyl diazoesters meets these requirements, providing silene equivalents in the form of silaoxetenes, that have appreciable room-temperature lifetimes. Most importantly, the results demonstrate that the acylsilene-to-silaoxetene isomerization is reversible at ambient temperature allowing these reagents to be combined with a range of α,β-unsaturated carbonyl compounds under very mild conditions. The resultant cyclic silenol ethers contain a high degree of functionality which offers considerable potential for further synthetic elaboration. In preliminary experiments simple treatment with fluoride afforded 1,5-dicarbonyl compounds in good yields. This overall sequence is therefore one in which the ‘silene’ has effectively functioned as a neutral enolate equivalent in the conjugate addition to chalcone thus further extending the silene neutral anion analogy.3b Efforts are currently underway to enhance this sequence: for example, silyl diazo precursors in which one or more of the trimethylsilyl groups can be replaced by other substituents, potentially affording alternative silene equivalents and cycloadducts. This requires careful control of migratory aptitude18 and the results of such studies will be reported in due course.
Acknowledgements
We thank the EPSRC (DTA studentship to M. C.), AstraZeneca and the Swedish Research Council (VR) for financial support of this work (CASE award to M. C.); the EPSRC Mass Spectrometry Service for accurate mass determinations, Dr A. M. Kenwright (University of Durham) for assistance with NMR experiments and Drs M. Jones and J. A. Mosely (University of Durham) for mass spectra.
Notes and references
-
(a) H. Ottosson and A. M. Eklöf, Coord. Chem. Rev., 2008, 252, 1287–1314 CrossRef CAS;
(b) R. West, Polyhedron, 2002, 21, 467–472 CrossRef CAS;
(c)
T. Müller, W. Ziche and N. Auner, in The Chemistry of Organic Silicon Compounds, vol. 2, ed. Z. Rappoport and Y. Apeloig, Wiley, Chichester, 1998, p. 857 Search PubMed;
(d) A. G. Brook and M. A. Brook, Adv. Organomet. Chem., 1996, 39, 71–158 CrossRef CAS;
(e)
G. Raabe and J. Michl, in The Chemistry of Organosilicon Compounds, ed. S. Patai and Z. Rappoport, J. Wiley and Sons Ltd., Chichester, 1989, pp. 1044–1102 Search PubMed;
(f) A. G. Brook and K. M. Baines, Adv. Organomet. Chem., 1986, 25, 1–44 CrossRef CAS.
-
(a) H. Ottosson and P. G. Steel, Chem.–Eur. J., 2006, 12, 1576–1585 CrossRef CAS;
(b) H. Ottosson, Chem.–Eur. J., 2003, 9, 4144–4155 CrossRef CAS.
-
(a) J. D. Sellars and P. G. Steel, Tetrahedron, 2009, 65, 5588–5595 CrossRef CAS;
(b) J. Bower, M. R. Box, M. Czyzewski, A. E. Goeta and P. G. Steel, Org. Lett., 2009, 11, 2744–2747 CrossRef CAS;
(c) R. D. C. Pullin, J. D. Sellars and P. G. Steel, Org. Biomol. Chem., 2007, 5, 3201–3206 RSC; N. J. Hughes, R. D. C. Pullin, M. J. Sanganee, J. D. Sellars, P. G. Steel and M. J. Turner, Org. Biomol. Chem., 2007, 5, 2841–2848 Search PubMed;
(d) J. D. Sellars and P. G. Steel, Org. Biomol. Chem., 2006, 4, 3223–3224 RSC;
(e) J. D. Sellars, P. G. Steel and M. J. Turner, Chem. Commun., 2006, 2385–2387 RSC;
(f) M. B. Berry, R. J. Griffiths, M. J. Sanganee, P. G. Steel and D. K. Whelligan, Org. Biomol. Chem., 2004, 2, 2381–2392 RSC;
(g) M. J. Sanganee, P. G. Steel and D. K. Whelligan, Org. Biomol. Chem., 2004, 2, 2393–2402 RSC;
(h) M. B. Berry, R. J. Griffiths, M. J. Sanganee, P. G. Steel and D. K. Whelligan, Tetrahedron Lett., 2003, 44, 9135–9138 CrossRef CAS;
(i) M. B. Berry, R. J. Griffiths, J. A. K. Howard, M. A. Leech, P. G. Steel and D. S. Yufit, J. Chem. Soc., Perkin Trans. 1, 1999, 3645–3650 RSC;
(j) M. B. Berry, R. J. Griffiths, D. S. Yufit and P. G. Steel, Chem. Commun., 1998, 2155–2156 Search PubMed;
(k) A. S. Batsanov, I. M. Clarkson, J. A. K. Howard and P. G. Steel, Tetrahedron Lett., 1996, 37, 2491–2494 CrossRef CAS.
- W. Schlenck and J. Renning, Justus Liebigs Ann. Chem., 1912, 394, 221 Search PubMed.
- L. E. Gusel'nikov and M. C. Flowers, Chem. Commun., 1967, 864–865 RSC.
-
(a) O. L. Chapman, C. C. Chang, J. Kolc, M. E. Jung, J. A. Lowe, T. J. Barton and M. L. Tumey, J. Am. Chem. Soc., 1976, 98, 7844–7846 CrossRef CAS;
(b) M. R. Chedekel, M. Skoglund, R. L. Kreeger and H. Shechter, J. Am. Chem. Soc., 1976, 98, 7846–7848 CrossRef CAS;
(c) W. Ando, A. Sekiguchi, T. Hagiwara, T. Migita, V. Chowdhry, F. H. Westheimer, S. L. Kammula, M. Green and M. Jones, J. Am. Chem. Soc., 1979, 101, 6393–6398 CrossRef CAS;
(d) A. Sekiguchi and W. Ando, Chem. Lett., 1983, 871–874 CrossRef CAS;
(e) A. Sekiguchi and W. Ando, Organometallics, 1987, 6, 1857–1860 CrossRef CAS;
(f) H. Tanaka, M. Ichinohe and A. Sekiguchi, Chem. Lett., 2008, 37, 1246–1247 CrossRef CAS.
-
(a)
G. Maas, in The Chemistry of Organosilicon Compounds, vol. 2, ed. S. Patai, Z. Rappoport and Y. Apeloig, J. Wiley and Sons Ltd., Chichester, 1998, ch. 13, pp. 703–778 Search PubMed;
(b) S. P. Marsden and W. K. Pang, Chem. Commun., 1999, 1199–1200 RSC;
(c) A. Sekiguchi, T. Sato and W. Ando, Organometallics, 1987, 6, 2337–2341 CrossRef CAS;
(d) A. Sekiguchi, W. Ando and K. Honda, Tetrahedron Lett., 1985, 26, 2337–2340 CrossRef CAS;
(e) M. Alt and G. Maas, Chem. Ber., 1994, 127, 1537–1542 CrossRef CAS;
(f) M. Alt and G. Maas, Tetrahedron, 1994, 50, 7435–7444 CrossRef CAS;
(g) G. Maas, M. Gimmy and M. Alt, Organometallics, 1992, 11, 3813–3820 CrossRef CAS;
(h) G. Maas, M. Alt, K. Schneider and A. Fronda, Chem. Ber., 1991, 124, 1295–1300 CrossRef CAS;
(i) K. Schneider, B. Daucher, A. Fronda and G. Maas, Chem. Ber., 1990, 123, 589–594 CrossRef CAS;
(j) R. Bruckmann, K. Schneider and G. Maas, Tetrahedron, 1989, 45, 5517–5530 CrossRef;
(k) R. Bruckmann and G. Maas, Chem. Ber., 1987, 120, 635–641 CrossRef.
- M. P. Doyle, Prog. Inorg. Chem., 2001, 49, 113–168 CrossRef CAS.
-
(a) T. L. Morkin and W. J. Leigh, Acc. Chem. Res., 2001, 34, 129–136 CrossRef CAS;
(b) A. M. Eklöf and H. Ottosson, Tetrahedron, 2009, 65, 5521–5526 CrossRef;
(c) A. M. Eklöf, T. Guliashvili and H. Ottosson, Organometallics, 2008, 27, 5203–5211 CrossRef.
-
Gaussian 03, Revision C.02, M. J. Frisch et al., Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed. For complete reference see ESI†.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS;
(b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef.
- T. Allspach, H. Gumbel and M. Regitz, J. Organomet. Chem., 1985, 290, 33–39 CrossRef CAS.
- Prepared from phenyl tris(trimethylsilyl)silane ( M. J. Sanganee, P. G. Steel and D. K. Whelligan, J. Org. Chem., 2003, 68, 3337–3339 CrossRef CAS ) by a sequence involving treatment with KOtBu and then reaction of the resulting silyl potassium with the required electrophile, C. Marschner, Eur. J. Inorg. Chem., 1998, 221–226 CrossRef . For full details see ESI†.
-
Catalysts explored included CuOTf, CuI, Cu(OAc)2, Cu(acac)2, Cu(tfacac)2, Pd(OAc)2, Pd(acac)2, Rh2(OAc)4 and Rh2(tfa)4. The copper catalysts led to extensive decomposition and intractable mixtures of products whilst palladium and rhodium complexes led to the recovery of
starting materials.
-
(a) A. Sekiguchi and W. Ando, J. Am. Chem. Soc., 1984, 106, 1486–1487 CrossRef CAS;
(b) G. Maas, K. Schneider and W. Ando, J. Chem. Soc., Chem. Commun., 1988, 72–74 RSC.
- I. Bejan, D. Güclü, S. Inoue, M. Ichinohe, A. Sekiguchi and D. Scheschkewitz, Angew. Chem., Int. Ed., 2007, 46, 3349–3352 CrossRef CAS.
-
Me3Si is known to migrate preferentially in competition to Me under photochemical irradiation, see ref 7i, whilst under metal catalysed initiation, trialkylsilyldiazoacetates afford no evidence for silene formation See for example: S. P. Marsden and W.-K. Pang, Tetrahedron Lett., 1998, 39, 6077 CrossRef CAS; S. N. Kablean, S. P. Marsden and A. M. Craig, Tetrahedron Lett., 1998, 39, 5109 CrossRef; C. Bolm, A. Kasyan, K. Drauz, K. Günther and G. Raabe, Angew. Chem., Int. Ed., 2000, 39, 2288 CrossRef; P. Müller and F. Lacrampe, Helv. Chim. Acta, 2004, 87, 2848 CrossRef . However, to the best of our knowledge, participation of aryl groups in this process has not been described.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of computational and experimental procedures, cartesian coordinates of calculated structures and copies of NMR spectra. See DOI: 10.1039/c1sc00405k |
| ‡ Current address: The Beatson Institute for Cancer Research, Garscube Estate, Switchback Road, Bearsden, Glasgow, UK G61 1BD. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.