Band gap engineering in fluorescent conjugated microporous polymers†
Jia-Xing
Jiang
,
Abbie
Trewin
,
Dave J.
Adams
* and
Andrew I.
Cooper
*
Department of Chemistry and Centre for Materials Discovery, University of Liverpool, Crown Street, Liverpool, UK. E-mail: aicooper@liv.ac.uk; d.j.adams@liverpool.ac.uk
First published on 7th July 2011
Abstract
We report here the synthesis of conjugated microporous polymers (CMPs) based on pyrene building units. The networks are both microporous and highly luminescent. The emission colour and resulting band gap can be fine tuned by introducing different comonomers and by varying the monomer distribution (statistical versus alternating). These materials might find applications in organic electronics, photocatalysis, optoelectronics, or in sensing technologies.
Introduction
Microporous organic polymers have broad potential in applications such as molecular separation, storage and catalysis.1 There are several sub-classes of porous polymers: these include polymers of intrinsic microporosity,1a,2 hypercrosslinked polymers,3 covalent organic frameworks (COFs),4 and conjugated microporous polymers (CMPs).5 An important advantage of microporous organic polymers is the potential to introduce synthetically a range of useful chemical functionalities into the pores.6Conjugated organic polymers have been studied intensively because of their applications in organic electronics and optoelectronics. Conjugated polymers, such as polyphenylenevinylenes,7 polyfluorenes,8 polycarbazoles,9 polyaryleneethynylenes,10 and polyphenylenes,11 tend to be luminescent and semi-conducting, and are used in organic light-emitting-diodes (OLEDs) and organic solar cells. Microporous polymers can also exhibit extended π-conjugation in the form of CMPs,5,12 and luminescent chromophores can be introduced, either in main chain or in a side chain appended to the network. Weber and Thomas12c reported the first example of an amorphous, blue-light-emitting microporous polymer derived from a 9,9′-spirobifluorene monomer. Subsequently, Jiang et al.13 showed the first example of a crystalline blue luminescent and semiconducting COF which consists of alternate pyrene and triphenylene functionalities within a mesoporous hexagonal skeleton. Rosseinsky et al.14 reported the synthesis of luminescent metal–organic frameworks (MOFs) by using a pyrene-derived linker. Each of these discoveries suggests technological possibilities in areas such as OLEDs, sensors, solar energy conversion, and hybrid conjugated materials.
Pyrene is an efficient chromophore that has been studied widely in the area of OLEDs. Many polymers based on pyrene derivatives have been synthesized including oligopyrene,15 poly(2,7-pyrenylene)s,16 polypyrene copolymers,17 and polypyrene dendrimers.18 Most of these materials are grafted with flexible groups to render them soluble in common solvents—in general, these solubilizing functionalities combined with a strong tendency for interchain π-stacking could render the materials non-porous. Here, we present the synthesis of a novel class of highly luminescent nanoporous networks based on pyrene building blocks viaNi-catalyzed Yamamoto reactions12d of 1,3,6,8-tetrabromopyrene (TBrPy) with comonomers such as 1,4-dibromobenzene (DB) and 4,4′-dibromobiphenyl (DP). The resulting polypyrene networks show high levels of porosity (surface areas up to 1508 m2 g−1) combined with strong fluorescence. This allows band gap engineering in porous networks, as well as the synthesis of microporous polymers with colour-tunable fluorescence.
Results and discussion
The general synthetic route for the polymer and copolymer networks is shown in Scheme 1. Yamamoto polymerization has been shown recently to be an efficient method to produce highly porous homocoupled polymer19 and copolymer networks.12d Similar methods were used here to synthesize luminescent porous polypyrene networks. Reaction of TBrPy in a mixture of N,N-dimethylformamide (DMF), 1,5-cyclooctadiene, bis(1,5-cyclooctadine)nickel (0), and 2,2′-bipyridyl gave the homocoupled polymer, YPy, as a brown, insoluble powder. Equivalent reactions of TBrPy with the linear, ditopic linkers DB or DP gave copolymers YDBPy and YDPPy, respectively. The Yamamoto copolymerization yields statistical copolymers when a second functionalized monomer is added to the reaction mixture.12d That is, the network does not form a perfect alternating structure as depicted in the simple representations given in Scheme 1. All of the polymers were insoluble in common organic solvents and were also chemically stable: for example, with respect to aqueous solutions of acids and bases, such as HCl and NaOH.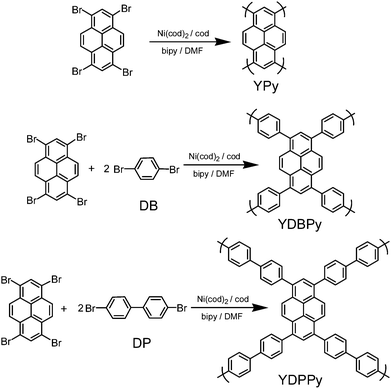 | ||
| Scheme 1 Synthetic routes to luminescent CMPs showing notional network structures. The actual structures of these statistical copolymers will be more complex than represented. | ||
Characterization of the networks by FT-IR spectroscopy, elemental analysis, and energy dispersive X-ray spectroscopy (EDX) confirmed the chemical identity of the samples. The absence of the C–Br stretching peak around 500 cm−1 in the FT-IR spectra demonstrated that most of the bromine functional group in the starting materials have been consumed by phenyl–phenyl coupling (see Supporting Information, Figure S1–S3†). Only a trace of bromine originating from unreacted end groups was detected by elemental analysis (0.2, 0.6 and 1.6 wt% for YPy, YDBPy and YDPPy, respectively). No nickel residue could be detected in the networks by EDX after exhaustive purification. Powder X-ray diffraction measurements revealed that all three polymer networks were amorphous in nature, as found for other Yamamoto coupled networks,12d,19a with the exception of PAF-119b,20 where some limited evidence for order was given based on broad peaks in the 5°–25° 2θ region. Thermogravimetric analysis indicated that all of the polymers were thermally stable up to 400 °C, and the materials only lost around 20 wt% of mass at 1000 °C (Fig. 1).
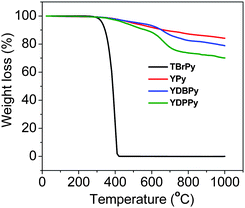 | ||
| Fig. 1 Thermogravimetric analysis (TGA) curves for three polymer networks and the pyrene monomer, TBrPy, recorded under a nitrogen atmosphere with a heating rate of 10 °C min−1. | ||
This high physicochemical stability can be explained by the rigid nature of these aromatic polymers, composed solely of strong carbon–carbon and carbon–hydrogen bonds. Scanning electron microscopy showed that YPy and YDBPy are produced as small spheres, which agglomerate and react to form larger structures. YDPPy shows a different texture with sheet-like structure on the micrometre scale (Supporting Information, Fig.S24–S26†).
The homocoupled polymer YPy is composed solely of pyrene units: like all of the networks presented here, this can be thought of as a contorted subset of a graphene sheet. However, unlike the 2-D, sheet-like structure of graphene,21 twisting between pyrene units is expected in these networks (Fig. 2a) by analogy with structural models for pyrene dendrimers18 and with X-ray crystal structures for analogous pyrene linkers in MOFs, such as 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy).14 This twisting would, in turn, be expected to inhibit pyrene aggregation which would both generate porosity and prevent fluorescence quenching in the solid state.
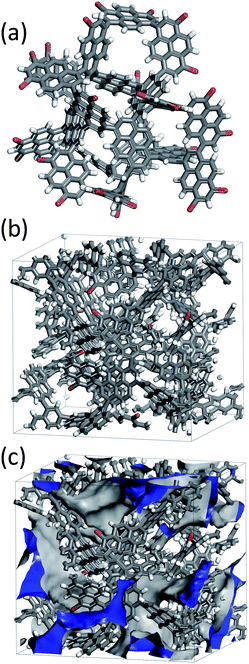 | ||
| Fig. 2 (a) A bromine-terminated fragment model used to construct the molecular simulations in (b, c); (b) An amorphous cell constructed from the YPy cluster; (c) A solvent-accessible surface area of 2016 m2 g−1, shown in blue, was calculated using a probe radius of 1.82 Å, the kinetic radius of N2. | ||
These materials are amorphous, and it is not possible to obtain an unambiguous 3-dimensional structure via X-ray methods, as for crystalline networks such as COFs.4,13 An atomistic model was therefore constructed for the YPy network using a cluster of 21 pyrene building units using the Materials Studio 5.0 package. The YPy cluster was fully optimized using the PCFF force field and charges calculated using the Gasteiger method in the Forcite module.22 The pyrene repeat units were held rigid but allowed to move relative to one another. As for the pyrene dendrimer model reported by Müllen et al.,18 the dihedral angle between adjacent pyrene repeat units was found to fall in one of three ranges depending on the position in the network (55–65°, 70–80°, and 85–95°). An amorphous cell with periodic boundary conditions was constructed: this was packed with the YPy-cluster and these cluster fragments were then linked together using additional pyrene units to form an infinite network. It was challenging in this model to replicate the low bromine content measured experimentally for the YPy network (0.2 wt. %) which equates to approximately 1 bromine per 150 repeat units. In the absence of any side reactions to cleave Br from the monomer, this is only possible if a highly condensed network is formed with very few end groups.20 Simulating this would have required a prohibitively large cell, requiring methodological advances in automation of the network construction. As such, a smaller cell comprising 55 pyrene repeat units was employed which yielded an artificially high Br end-group content of 5.5 wt. %. The experimentally determined absolute density (0.885 g cm−3) and micropore volume (0.357 cm3 g−1) for YPy (see gas sorption discussion, below) were used to calculate a target bulk density value of 0.67 g cm−3 for the simulation.3b The cell was optimised with the pyrene repeat units held rigid but allowed to move relative to each other. A simulated density of 0.71 g cm−3 was obtained, close to the experimental target. The solvent-accessible surface area calculated using a probe radius of 1.82 Å (the kinetic radius of N2) was 2016 m2 g−1. A relatively broad pore size distribution was found in the simulation, ranging from 7.5 to 16.0 Å with an average simulated pore diameter of 11.3 Å.
The porosity in the networks was investigated by nitrogen adsorption analysis. Fig. 3a shows the N2 adsorption and desorption isotherms for the resulting polymers. All polymer networks gave rise to N2 gas sorption isotherms with Type I character indicative of micropores,23 along with sorption at higher relative pressures and H3 hysteresis loops indicating a degree of mesoporosity. Hysteresis was observed persisting to low relative pressures for all of the three polymers, consistent with elastic deformations or swelling as a result of gas sorption.24
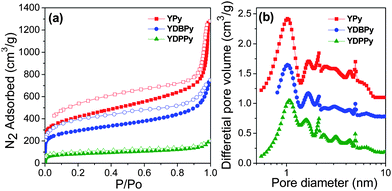 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms measured at 77.3 K (adsorption branch is labelled with filled symbols); (b) NL-DFT pore size distribution curves for the three polymers (see also Table 1). | ||
Fig. 3b shows the pore size distribution curves for the networks as calculated using the non-local density functional theory (NL-DFT). All polymers show a median pore diameter of around 1.1 nm and a relatively broad pore size distribution, consistent with the approximate values derived from the structural model presented in Fig. 2. The apparent Brunauer-Emmet-Teller (BET) surface areas were found to be 1508, 1069, and 303 m2 g−1 for YPy, YDBPy and YDPPy, respectively. This decreasing trend was attributed to the increased average linker length between the pyrene nodes in the networks which results from incorporating a second monomer (DB or DP). A similar trend was observed for CMPs synthesized via both Sonogashira–Hagihara coupling12b,25 and Yamamoto coupling,12d most likely because of increased interpenetration as the average strut length increases. The surface area for the YPy network (1508 m2 g−1) is higher than most CMPs produced so far which have extended conjugation: that is, excluding networks such as PAF-1 derived from tetrahedral monomers19 where this conjugation is broken by the sp3-hybridized carbon node. The measured apparent BET surface area for YPy is broadly consistent with the calculated solvent-accessible surface area obtained from the molecular simulation in Fig. 2.
All polymers show strong solid state fluorescence with emission colours ranging from red (YPy) to orange (YDPPy) to yellow (YDBPy) (Fig. 4a–c). The absence of fluorescence quenching in the solid state is consistent with the contorted porous network structure suggested by the simulation in Fig. 2. Indeed, the fluorescence in YPy can be almost totally quenched if the porous network is imbibed with a solution of pyrene in tetrahydrofuran (THF) (Supporting Information, Figure S18†).
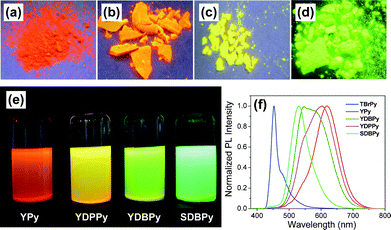 | ||
| Fig. 4 Photographs of the polymers under irradiation with UV light (λexcit = 365 nm) in the solid state, (a) YPy, (b) YDPPy, (c) YDBPy, and (d) SDBPy; (e) Photographs of suspensions of the polymers in THF (10 mg/10 mL); (f) Photoluminescent spectra of the monomer TBrPy and the resulting polymers measured in solid state powder (λexcit = 360 nm). | ||
Suspensions of the polymers in solvents such as THF show blue-shifted emissions ranging from orange to yellow to green for YPy, YDPPy and YDBPy, respectively (Fig. 4e). Essentially no fluorescence remained in the filtrate after filtration to remove the insoluble network particles (Fig. S14†), confirming that we had successfully removed any soluble monomers or oligomers from the networks. Fig. 4f shows the photoluminescent emission spectra of the monomer TBrPy in comparison with four polymer networks in solid state at room temperature. The emission for TBrPy is blue shifted with respect to the networks, with a maximum characteristic emission at 452 nm and a shoulder around 475 nm. By contrast, the homocoupled polymer network formed from TBrPy, YPy, shows a saturated red emission at around 620 nm as a result of the extended conjugation in the polymer. The copolymer YDPPy shows an orange emission peak at 602 nm, while YDBPy shows a broader, green emission spectrum with two main peaks centred at 545 and 582 nm. Müllen et al.18 observed that the fluorescence band red shifts as a function of the number of pyrene units in 1,3,6,8-conjugated polypyrene dendrimers, again because of increased conjugation length. This implies that the degree of polymerization in our polymers is high, in agreement with the low bromine contents in the samples.
Two emission peaks were observed for both of the copolymers YDPPy (602 and 545 nm) and YDBPy (545 and 582 nm; Fig. 4f), while YPy showed a more symmetrical emission peak centred at 620 nm. We suggest that this is due to the presence of a distribution of chromophores in the copolymer networks, resulting from the statistical distribution of monomer sequences (i.e., for YDBPy – TBrPy-TBrPy, TBrPy-DB, DB-DB sequences, etc.) To investigate this, we synthesized another copolymer, SDBPy, via Suzuki copolymerization of TBrPy and 1,4-benzene diboronic acid to produce a copolymer with an unambiguous alternating structure, as shown in Scheme 1. SDBPy showed high porosity with a surface area of 976 m2 g−1, similar to the statistical copolymer YDBPy (1069 m2 g−1). Moreover, SDBPy showed a strong green emission with a single, much narrower emission band centred at 530 nm (Fig. 4d and f). These data show that it is possible to fine tune the electronic structure and therefore the band gap (Table 1) in CMP networks by judicious choice of comonomer, much as we demonstrated that surface area was continuously tunable in poly(aryleneethynylene) copolymers.12b,25
Conclusions
In conclusion, we demonstrate here an approach to produce CMPs based on pyrene building units. All of the networks are microporous and highly luminescent, and the emission colour and resulting band gap can be fine tuned by introducing different comonomers and by varying the monomer distribution (statistical versus alternating). These materials could find a wealth of applications in organic electronics, optoelectronics, and in sensing technologies. For example, these materials can be considered as nascent bulk heterojunctions where one of the phases (the micropores) consists of vacant space and might be filled with a wide range of dopants or second phases. Moreover, the observed band gaps in these polymers are in the range which is relevant for applications such as photochemical water splitting,26 expressed here in materials with large distributed surfaces.Acknowledgements
The authors gratefully acknowledge EPSRC for funding (EP/F057865/1 and EP/H000925). AIC is a Royal Society Wolfson Merit Award holder. We thank Dr T. Hasell for assistance with SEM and Mr R. Clowes for gas sorption measurements.References
- (a) N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163 CrossRef CAS; (b) F. Svec, J. Germain and J. M. J. Fréchet, Small, 2009, 5, 1098 CrossRef; (c) J.-X. Jiang and A. I. Cooper, Top. Curr. Chem., 2010, 293, 1 CrossRef CAS; (d) A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328 CrossRef CAS.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230 RSC.
- (a) M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193 CrossRef CAS; (b) C. D. Wood, B. Tan, A. Trewin, H. J. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stockel and A. I. Cooper, Chem. Mater., 2007, 19, 2034 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef.
- (a) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS; (b) A. I. Cooper, Adv. Mater., 2009, 21, 1291 CrossRef CAS.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS.
- J. F. Morin, M. Leclerc, D. Ades and A. Siove, Macromol. Rapid Commun., 2005, 26, 761 CrossRef.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS.
- (a) A. J. Berresheim, M. Muller and K. Müllen, Chem. Rev., 1999, 99, 1747 CrossRef CAS; (b) A. C. Grimsdale and K. Müllen, Adv. Polym. Sci., 2006, 199, 1 CrossRef CAS.
- (a) J.-X. Jiang, F. Su, H. J. Niu, C. D. Wood, N. L. Campbell, Y. Z. Khimyak and A. I. Cooper, Chem. Commun., 2008, 486 RSC; (b) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710 CrossRef CAS; (c) J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334 CrossRef CAS; (d) J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426 CrossRef CAS; (e) L. Chen, Y. Yang and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 9138 CrossRef CAS; (f) J. X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. L. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072 CrossRef CAS; (g) R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173 RSC.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. L. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826 CrossRef CAS.
- K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119 CrossRef CAS.
- (a) L. T. Qu and G. Q. Shi, Chem. Commun., 2004, 2800 RSC; (b) X. G. Li, Y. W. Liu, M. R. Huang, S. Peng, L. Z. Gong and M. G. Moloney, Chem.–Eur. J., 2010, 16, 4803 CAS.
- S. I. Kawano, C. Yang, M. Ribas, S. Baluschev, M. Baumgarten and K. Müllen, Macromolecules, 2008, 41, 7933 CrossRef CAS.
- J. A. Mikroyannidis, Synth. Met., 2005, 155, 125 CrossRef CAS.
- T. M. Figueira-Duarte, S. C. Simon, M. Wagner, S. I. Drtezhinin, K. A. Zachariasse and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 10175 CrossRef CAS.
- (a) J. R. Holst, E. Stöckel, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8531 CrossRef CAS; (b) T. Ben, H. Ren, S. Q. Ma, D. P. Cao, J. H. Lan, X. F. Jing, W. C. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457 CrossRef CAS.
- A. Trewin and A. I. Cooper, Angew. Chem., Int. Ed., 2010, 49, 1533 CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- (a) J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219 CrossRef CAS; (b) J. R. Maple, M. J. Hwang, T. P. Stockfisch, U. Dinur, M. Waldman, C. S. Ewig and A. T. Hagler, J. Comput. Chem., 1994, 15, 162 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880 CrossRef CAS.
- J.-X. Jiang, A. Trewin, F. B. Su, C. D. Wood, H. J. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658 CrossRef CAS.
- M. G. Schwab, M. Hamburger, X. L. Feng, J. Shu, H. W. Spiess, X. C. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, IR data, SEM, XRD, further fluorescence and gas sorption data. See DOI: 10.1039/c1sc00329a |
| This journal is © The Royal Society of Chemistry 2011 |