Chemical control of interstitial iron leading to superconductivity in Fe1+xTe0.7Se0.3†
Efrain E.
Rodriguez
a,
Christopher
Stock
ab,
Ping-Yen
Hsieh
ac,
Nicholas P.
Butch
d,
Johnpierre
Paglione
d and
Mark A.
Green
*ac
aNIST Center for Neutron Research, NIST, Gaithersburg, MD 20899, USA. E-mail: mark.green@nist.gov
bIndiana University, 2401 Milo B. Sampson Lane, Bloomington, Indiana 47408, USA
cDepartment of Materials Science and Engineering, University of Maryland, College Park, 20742, USA
dCenter for Nanophysics and Advanced Materials, University of Maryland, College Park, MD 20742, USA
First published on 7th July 2011
Abstract
Although it possesses the simple layered topology of the tetragonal anti-PO structure, the Fe(Te,Se) series has a complex structural and magnetic phase diagram that is dependent on composition and occupancy of a secondary interstitial Fe site. Here we show that superconductivity in Fe1+xTe0.7Se0.3 is enhanced by topotactic deintercalation of the interstitial iron with iodine, demonstrating the competing roles of the two iron positions. We follow the evolution of the structure and magnetic properties as a function of interstitial iron. Powder neutron diffraction reveals a flattening of the Fe(Te,Se)4 tetrahedron on Fe removal and an unusual temperature dependence of the lattice parameters that increases strongly below 150 K along with lattice strain. Inelastic neutron scattering shows gapless paramagnetic scattering evolves into a gapped excitation at 6 meV on removal of interstitial iron. This work highlights the robustness of the superconductivity across different Fe(Te,Se) compositions and geometries.
Introduction
Iron based high temperature superconductors offer new opportunities to establish the interplay between magnetism, composition and electronic properties.1 Several structural families have now been established since the discovery of superconductivity at 26 K in LaO1−xFxFeAs.2 These iron pnictide systems require additional cations to provide charge balancing, which is not necessary in the structurally simpler superconducting iron chalcogenide series, FeX (X = Te, Se, S).3–5 Within this series, stoichiometric FeSe is orthorhombic and superconducting at 8.5 K,3,6 which increases to 36.7 K under high pressure.7–9 The work on stoichiometric FeSe,6 as with studies on Li1−yFe1+yAs,10 demonstrates the very rapid suppression of superconductivity with composition. Fe1+xTe can only be synthesized with large amounts of interstitial iron. This additional iron is known to greatly affect its structural and magnetic properties. For example, for x > 0.12 the tetragonal structure transforms to a orthorhombic one with an incommensurate magnetic structure at low temperature, in contrast to the monoclinic symmetry and a commensurate magnetic structure for lower iron levels.11 Using conventional solid state chemistry methodology, it was reported that Fe1+xTe can be formed at least over a range from Fe1.076(2)Te to Fe1.141(2)Te.11 Additional control of the concentration can be achieved by deintercalation of the interstitial iron with iodine, transforming Fe1.18(5)Te to Fe1.042(5)Te.12Previous studies on Fe1+xTe1−ySey have noted that optimal superconductivity has been limited to compositions around y ∼ 0.5.13 However, a strong correlation has also been reported between anion composition and the amount of interstitial iron present for both the Fe(Te,Se)13 and Fe(Te,S)14 phase diagrams. The Se or S substitution reduces the lattice volume, imposes chemical pressure, and suppresses the amount of interstitial iron present. It is therefore unclear whether the particular compositions that are noted for their superconductivity are a result of optimal Te![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Se ratios or whether the presence of interstitial iron, despite occupancies of only a few percent, is playing the prevailing role on the electronic properties. Previous attempts to study the effect of interstitial iron on the structural and magnetic properties of the Fe1+x(Te,Se) series have shown that interstitial iron may be detrimental to the superconductivity.15–17 However, the interstitial iron content is coupled with the Te:Se ratio, and all previous attempts to synthesize samples with different iron content also result in variation of the Te:Se ratio, as evidenced by variation of lattice parameters.16,17 Here, we take a single batch of Fe1.048(2)Te0.7Se0.3 and topotactically deintercalate different amounts of excess iron with iodine, producing a range of compositions at a fixed Te:Se ratio, where the lattice parameter, c, varies by only 0.0037(2) Å across all compositions. We show that the secondary interstitial iron is the critical parameter and superconductivity in other Te:Se ratios can be artificially produced by removal of the interstitial iron. The Fe1+xTe0.7Se0.3 composition with 70:30 ratio of Te:Se was specifically chosen as it has been previously reported as having both a very high18 and very low13,19 superconducting volume fraction.
:Se ratios or whether the presence of interstitial iron, despite occupancies of only a few percent, is playing the prevailing role on the electronic properties. Previous attempts to study the effect of interstitial iron on the structural and magnetic properties of the Fe1+x(Te,Se) series have shown that interstitial iron may be detrimental to the superconductivity.15–17 However, the interstitial iron content is coupled with the Te:Se ratio, and all previous attempts to synthesize samples with different iron content also result in variation of the Te:Se ratio, as evidenced by variation of lattice parameters.16,17 Here, we take a single batch of Fe1.048(2)Te0.7Se0.3 and topotactically deintercalate different amounts of excess iron with iodine, producing a range of compositions at a fixed Te:Se ratio, where the lattice parameter, c, varies by only 0.0037(2) Å across all compositions. We show that the secondary interstitial iron is the critical parameter and superconductivity in other Te:Se ratios can be artificially produced by removal of the interstitial iron. The Fe1+xTe0.7Se0.3 composition with 70:30 ratio of Te:Se was specifically chosen as it has been previously reported as having both a very high18 and very low13,19 superconducting volume fraction.
Results and discussion
A powder sample of nominal composition, Fe1.05Te0.7Se0.3, was synthesized by a solid state reaction of the constituent elements at 700 °C under vacuum. The energy dispersive X-ray (EDX) technique was found not to be sufficiently accurate to determine the occupancy of the iron interstitial sites. It has been reported that the occupancy values obtained for bulk single crystal X-ray diffraction determination and EDX measurements, which is a surface probe, yield different values.13 This implies that the stability of the excess iron is less on the surface and the iron adopts more locations in the bulk of the sample. Single crystal X-ray diffraction and powder neutron diffraction were found to give consistent and reliable values for the average composition. Samples of Fe1+xTe0.7Se0.3 were, therefore, characterized by high-resolution powder neutron diffraction using the BT1 diffractometer at NIST. For composition analysis, the samples were cooled to 100 K to reduce the thermal factors that can correlate with the occupancy values, and measured using the Cu 311 monochromator at the high 90° take-off angle, giving λ = 1.5401 Å. Temperature dependence of the lattice parameters and isotropic strain were performed using the Ge 311 monochromator at a 75° take-off angle, giving a wavelength, λ = 2.0787 Å. The actual composition was determined to be Fe1.048(2)Te0.7Se0.3.The composition Fe1.048(2)Te0.7Se0.3 was then divided into 4 batches, three of which were exposed to different levels of I2 vapour at 200 °C in an evacuated glass ampoule, which topotactically deintercalates the excess iron.12 The samples were subsequently washed with methanol to remove the FeI2 formed in the reaction, sonicated and centrifuged. A small (<1%) impurity of FeTe2 were present in samples that have been reacted with I2. Powder neutron diffraction at λ = 1.5401 Å determined the compositions of these to be Fe1.033(2)Te0.7Se0.3, Fe1.018(2)Te0.7Se0.3 and Fe1.009(3)Te0.7Se0.3. All refinements, which were carried out with the FULLPROF suite,20 gave the ratio of Te:Se to be 70:30 within the accuracy of the experiments, therefore this parameter was not included further in the refinements. A summary of the structural parameters for all four compositions at 100 K is given in Table 1.
| Fe1.009(3)Fe0.7Se0.3100 K | Fe1.018(2)Fe0.7Se0.3100 K | Fe1.033(2)Fe0.7Se0.3100 K | Fe1.048(2)Fe0.7Se0.3100 K | |
|---|---|---|---|---|
| Tez | 0.2136(7) | 0.2127(5) | 0.2109(4) | 0.2101(3) |
| Sez | 0.262(1) | 0.2607(9) | 0.2600(7) | 0.2614(6) |
| Fe2z | 0.72(2) | 0.72(1) | 0.749(5) | 0.774(3) |
| Fe2 occ | 0.009(3) | 0.018(2) | 0.033(2) | 0.048(2) |
| a/Å | 3.80197(4) | 3.80166(2) | 3.79971(2) | 3.79984(2) |
| c/Å | 6.08062(9) | 6.07568(6) | 6.07990(5) | 6.08386(4) |
| V/Å3 | 87.895(2) | 87.809(1) | 87.780(1) | 87.843(1) |
| R p | 4.85 | 5.05 | 5.00 | 4.12 |
| wRp | 6.99 | 6.54 | 6.83 | 5.72 |
| χ 2 | 2.04 | 1.38 | 1.76 | 1.77 |
| Fe1.009(3)Fe0.7Se0.37 K | Fe1.018(2)Fe0.7Se0.37 K | Fe1.033(2)Fe0.7Se0.37 K | Fe1.048(2)Fe0.7Se0.37 K | |
|---|---|---|---|---|
| Tez | 0.2155(7) | 0.2126(4) | 0.2106(4) | 0.2107(3) |
| Sez | 0.262(1) | 0.2615(7) | 0.2609(7) | 0.2611(5) |
| Fe2z | 0.72(2) | 0.709(8) | 0.763(5) | 0.776(2) |
| Fe2 occ | 0.009(—) | 0.018(—) | 0.033(—) | 0.048(—) |
| a/Å | 3.80429(4) | 3.80184(2) | 3.80023(2) | 3.80136(2) |
| c/Å | 6.06688(9) | 6.07415(5) | 6.07704(5) | 6.07420(4) |
| V/Å3 | 87.804(2) | 87.796(1) | 87.763(1) | 87.774(1) |
| R p | 4.73 | 4.19 | 5.07 | 3.70 |
| wRp | 6.85 | 5.55 | 6.90 | 5.32 |
| χ 2 | 1.91 | 1.60 | 1.78 | 1.82 |
| T c/K | 14.5 | 14.3 | 13.8 | 13.3 |
The c lattice parameter varies smoothly with Se incorporation into Fe1+xTe4, and which therefore offers a secondary approach to evaluate the composition. In our samples, the c parameter under ambient conditions refined to be 6.1165(1) Å, 6.11974(7) Å, 6.12021(7) Å and 6.12021(12) Å for Fe1.048(2)Te0.7Se0.3, Fe1.033(2)Te0.7Se0.3, Fe1.018(2)Te0.7Se0.3 and Fe1.009(3)Te0.7Se0.3, respectively. This represents a maximum change of c = 0.0037(2) between all phases, confirming that the interstitial iron has a very minor effect on the lattice parameter, which are almost wholly controlled by the composition. Producing different iron concentrations by low temperature deintercalation of a single Fe1+xTe1−ySey batch, eliminates Te:Se composition variation effects, which are evident in experiments that attempt to synthesize different compositions by direct reaction of elements with varying nominal composition. For example, the iodine deintercalation samples have a variation of the c lattice parameters that is 5 times less than those obtained using direct reactions.16,17 In addition, studies have also demonstrated that experimentally determined excess iron composition can be identical irrespective of the nominal composition used.17 This iodine deintercalation technique therefore allows for accurate experiments to be performed on the variation of Fe content at a fixed Te:Se ratio.
Fig. 1, panel a, shows the two iron locations in the anti-PbO structure of Fe1+xTe0.7Se0.3. The fully occupied iron, shaded orange, forms the main Fe(Te,Se)4 tetrahedron that is edge shared to form two dimensional layers. The nearest neighbour van der Waals bonded layers are shifted by (½ ½) in the ab plane. The partially occupied Fe sites, which in the case of Fe1+xFe0.7Se0.3 can adopt occupancies of up to ∼5%, sits directly above, and bonded to, the (Te, Se) split site in the layer below. It is further bonded to four other (Te, Se) sites within the ab plane to form a square pyramidal arrangement. Magnetization measurements, using a commercial SQUID magnetometer, were performed under zero-field cooled conditions on the four variable iron concentrations, Fe1.048(2)Te0.7Se0.3, Fe1.033(2)Te0.7Se0.3, Fe1.018(2)Te0.7Se0.3 and Fe1.009(3)Te0.7Se0.3 and are shown in Fig. 1, panel b. Fe1.048(2)Te0.7Se0.3 showed extremely low superconducting volume fractions. The extent of the superconducting volume fraction steadily increases with the removal of the interstitial iron, establishing a direct association between the two. The iodine deintercalation procedure is a topotactic technique that is done at very low temperatures, which are much below the synthesis temperature, thereby ruling out any possible changes to the actual Fe1+xTe0.7Se0.3 framework. In addition, the superconducting transition temperature is found to increase from 13.3 K to 14.5 K as a function of interstitial iron content (see Table 1). The value of 14.5 K is similar to the highest value for the superconducting transition temperature found in the Fe(Te,Se) series, and suggests a common origin for superconductivity across the series.
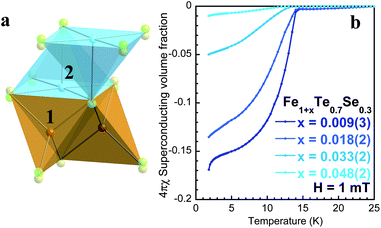 | ||
| Fig. 1 Panel a: Structure of Fe1+xTe0.7Se0.3, highlighting the two distinct iron locations; 1 forms the edge shared tetrahedral layers, while the partially occupied (< 5%) square pyramidal site 2 lies within the (Te, Se) plane. The Te and Se adopt very different positions along the z axis, creating a split site anion distribution. Panel b: Interstitial iron in Fe1+xTe0.7Se0.3 is the dominant factor controlling the balance between superconductivity and localized magnetism, as measure by a commercial SQUID magnetometer. | ||
Fig. 2, panel a and b, shows the lattice parameter as a function of temperature for the four compositions, as obtained from Rietveld refinements of powder neutron diffraction data. Each of the materials undergo unusual expansion of the ab plane below ∼125 K, whilst the c parameter shrinks in a typical linear fashion. Similar anisotropic thermal expansion has been previously reported in other members of the Fe1+x(Te,Se) series.21–23 The isotropic strain of the system, shown in Fig. 2 panel c, obtained from the peak shape parameters of the refinements, increases greatly below 150 K. The increase in both the lattice parameter and strain below 150 K suggests that the expansion in ab, which is directly related to the primary Fe–Fe distance, is a result of microstrain within the lattice. Analysis of the peak broadening (see supplementary information) ruled out the strain to be a result of a small orthorhombic distortion or even orthorhombic strain on a tetragonal lattice. Therefore the strain results from structural effects accumulating from the stacking of the layers and the disorder caused by the mixed Te and Se sites that have very different Fe–Te and Fe–Se bond distances. It is interesting to note that there is an overall increase in the ab plane on reduction of the interstitial iron, reflecting the attractive bonding involving the interstitial iron within the van der Waals layers. The distance between the layers is almost identical for all compositions. A graphical comparison of the effect of iron deintercalation on the structure is given in Fig. 3 panel a.
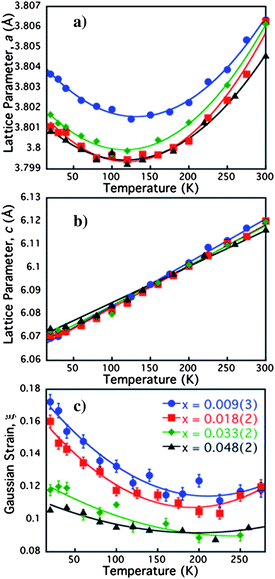 | ||
| Fig. 2 Panel a: Deintercalation of iron expands the ab lattice parameters which all also show an initial decrease on cooling from ambient conditions to 100 K, followed by an expansion to base whereas, Panel b: the c parameter is not greatly influenced by composition and shows a typical linear contraction with temperature. Panel c: Isotropic Gaussian strain within the lattice increases on both deintercalation of interstitial iron and on cooling. Error bars are shown for all figures and represent one standard deviation. | ||
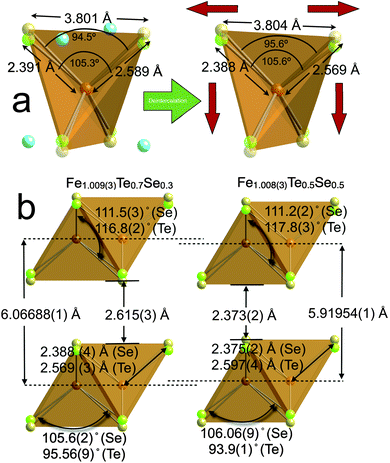 | ||
| Fig. 3 Panel a: Comparison of the structures at 7 K of Fe1+xTe0.7Se0.3 before and after removal of the interstitial iron with iodine, showing the deintercation process increases the Fe–Fe distances while squashing the Fe tetrahedron in the c direction. Panel b: Comparison of the structures of Fe1.009(3)Te0.7Se0.3 with Fe1.008(3)Te0.5Se0.5. The greatly increased c parameter in the 70:30 over the 50:50 composition of around 0.15 Å, combined with the collapse of the Fe(Te0.7Se0.3) tetrahedron demonstrated in the less acute Te–Fe–Te bond angles and shorter Fe–Te bond distances, results is a significantly increased interlayer separation of ∼0.24 Å. The superconducting Tc is unaffected by these structural changes. | ||
As a comparison with Fe1.009(3)Te0.7Se0.3, we determined by powder neutron diffraction, the structure of Fe1.008(3)Te0.48(1)Se0.52(2) and find some stark contrasts between their structures. The former has lattice parameters of a = 3.80429(4) Å and c = 6.06688(9) Å at 5 K, whereas the latter possesses lattice parameters of a = 3.79496(5) Å and c = 5.91966(12) Å at 5 K. The Fe–Fe distance is directly related to the lattice parameter, such that dFe–Fe = a/√2. Therefore, the small change in a of ∼0.01 Å, suggests that the Fe–Fe distances within the ab plane are relatively insensitive to the anion composition. Similarly, the Fe–Te and Fe–Se bond lengths and angles are relatively unchanged between for the two compositions, and remain almost identical to the values found for their parent Fe1+xTe and FeSe structures. In contrast, significant differences in bond distances along the c direction are observed; the 70:30 composition has significant shrinkage in the Fe tetrahedron or intralayers, but an increase between the van der Waals gap or interlayer spacing of ∼0.24 Å, which results in an overall increase in c of ∼0.15 Å, when compared with the 50:50 composition. As the superconducting transition temperatures are very similar in both compositions at ∼14.5 K, this confirms that the layer distances, and parameters related to this distance such as anion height, are not critical parameters in controlling the superconductivity. A detailed comparison of the two Fe environments is given in Fig. 3, panel b.
To understand the effects of deintercalation on the magnetic fluctuations in Fe1+x (Te,Se), we performed inelastic neutron scattering measurements; the results are summarised in Fig. 4. The sample with the lowest concentration of excess iron and largest superconducting volume fraction shows a distinct feature centred at a |Q| ≈ 1.4 Å−1 and with an energy gap between 6 and 8 meV. This excitation has previously been observed below the superconducting transition temperature in the compositions close to FeTe0.5Se0.5 and is thought to be associated with the superconducting state.1,19,24,25 As the interstitial iron concentration is increased, paramagnetic fluctuations fill in the energy gap, starting from |Q| ≈ 0.9 Å−1 close to the elastic line and dispersing towards the position of the gapped excitation (Fig. 4). For the sample with the maximum amount of excess iron, Fe1.048(2)Te0.7Se0.3, this paramagnetic scattering completely overwhelms any gapped excitation that may have been present. The inelastic spectra clearly indicate that small changes in the interstitial iron concentration, even by a few percent, are critical to the electronic and magnetic properties of the Fe1+x(Te,Se) superconductors.
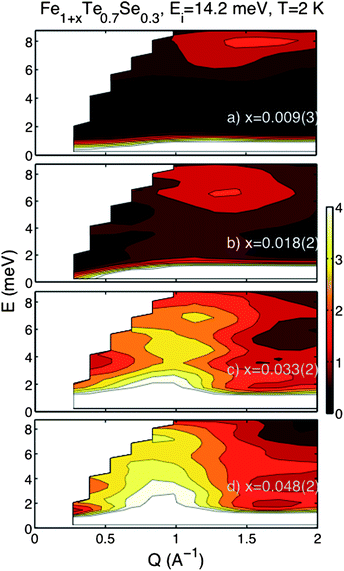 | ||
| Fig. 4 Evolution of the excitation spectrum in Fe1+xTe0.7Se0.3 at 2 K, showing the gapped excitation between 6–8 meV for x = 0.009(3) disappearing to gapless paramagnetic scattering on increase of interstitial iron to x = 0.048(2). | ||
There is substantial evidence pointing towards a direct coupling between antiferromagnetism and superconductivity in iron based superconductors. Mostly notably, strong magnetic collective excitations have been observed in both Ba0.6K0.4Fe2As226 and Fe(Te,Se)19,24,25 systems and have been thought to directly result from the presence of an electronic superconducting energy gap. The collective mode has been found to be gapped and to draw spectral weight from lower energies in these systems, conserving sum rules required in neutron scattering. Furthermore, inelastic neutron studies of other superconductors where magnetism is thought to play an important role, such as CeCu2Si2,27CeCoIn5,28 and YBa2Cu3O6.5,29 there is a clear shift in spectral weight from the gapped excitation to gapless magnetic fluctuations as these systems become non-superconducting. Therefore, the gapped fluctuations present in Fig. 4 point towards strong evidence that superconductivity and magnetism are directly coupled in these systems. For samples which are not bulk superconductors, the gapped magnetic fluctuations are replaced by gapless paramagnetic fluctuations. Our results demonstrate a direct coupling between superconductivity and antiferromagnetism and, most importantly, the iron doping on the interstitial site. These results also demonstrate a change in the wavevector of fluctuations with doping on the interstitial site. In previous neutron scattering experiments, the gapped excitation has been found to occur at a wavevector of Q = [½, ½] within the ab plane; this ordering corresponds to antiferromagnetic coupling along a diagonal of the Fe square sublattice. However, the magnetic ordering within Fe1+xTe occurs with a Q = [½, 0], or antiferromagnetic coupling along one side of the Fe square sublattice.11 While the data is powder averaged, the wavevectors are consistent with previous single crystal work, which demonstrates that the magnetic excitations change wavevector from [½ 0] to [½ ½] in the presence of superconductivity.18,19 Indeed, some density functional theory calculations studies have shown that the interstitial iron plays a crucial role on whether [½, ½]- or [½, 0]-type magnetic interactions will dominate.30 Thus, our results further advance the theory that magnetic interactions along Q = [½, 0], promoted by the interstitial iron sites, are antagonistic to superconductivity in the Fe1+x(Se,Te) series.
Conclusions
We have performed an accurate study of the effects of interstitial iron on the structure and superconductivity of Fe1+xTe1−ySey, and established that superconductivity can occur at different compositions and geometry as long as sufficient interstitial iron, which destroys superconductivity, is removed. This highlights the critical role of the iron concentrations and not Te:Se ratio on the electronic structure. A comparison between the two superconducting compositions of Fe1.009(3)Te0.7Se0.3 and Fe1.008(3)Te0.48(1)Se0.52(2) revealed some notable differences between their two structure. Nevertheless they show similar superconducting transition temperatures that demonstrates the robustness of superconductivity in the Fe(Te,Se) series. It will be interesting to evaluate the full phase diagram to establish whether Fe1+xTe or composition close to Fe1+xTe can support superconductivity given sufficient removal of interstitial iron.
Notes and references
- J. Paglione and R. L. Greene, Nat. Phys., 2010, 6, 645 CrossRef CAS
.
- Y. Kamihara, T. Watanabe, M. Hirano and H. Hosono, J. Am. Chem. Soc., 2008, 130, 3296 CrossRef CAS
- F. C. Hsu, J. Y. Luo, K. W. Yeh, T. K. Chen, T. W. Huang, P. M. Wu, Y. C. Lee, Y. L. Huang, Y. Y. Chu, D. C. Yan and M. K. Wu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14262–14264 CrossRef CAS
- K. W. Yeh, T. W. Huang, Y. L. Huang, T. K. Chen, F. C. Hsu, P. M. Wu, Y. C. Lee, Y. Y. Chu, C. L. Chen, J. Y. Luo, D. C. Yan and M. K. Wu, Europhys. Lett., 2008, 84, 37002 CrossRef
- Y. Mizuguchi, F. Tomioka, S. Tsuda, T. Yamaguchi and Y. Takano, Appl. Phys. Lett., 2009, 94, 012503 CrossRef
- T. M. McQueen, Q. Huang, V. Ksenofontov, C. Felser, Q. Xu, H. Zandbergen, Y. S. Hor, J. Allred, A. J. Williams, D. Qu, J. Checkelsky, N. P. Ong and R. J. Cava, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 014522 CrossRef
- Y. Mizuguchi, F. Tomioka, S. Tsuda, T. Yamaguchi and Y. Takano, Appl. Phys. Lett., 2008, 93, 152505 CrossRef
- S. Medvedev, T. M. McQueen, I. A. Troyan, T. Palasyuk, M. I. Eremets, R. J. Cava, S. Naghavi, F. Casper, V. Ksenofontov, G. Wortmann and C. Felser, Nat. Mater., 2009, 8, 630–633 CrossRef CAS
- S. Margadonna, Y. Takabayashi, Y. Ohishi, Y. Mizuguchi, Y. Takano, T. Kagayama, T. Nakagawa, M. Takata and K. Prassides, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80 CrossRef CAS
- M. J. Pitcher, T. Lancaster, J. D. Wright, I. Franke, A. J. Steele, P. J. Baker, F. L. Pratt, W. T. Thomas, D. R. Parker, S. J. Blundell and S. J. Clarke, J. Am. Chem. Soc., 2010, 132, 10467–10476 CrossRef CAS
- W. Bao, Y. Qiu, Q. Huang, M. A. Green, P. Zajdel, M. R. Fitzsimmons, M. Zhernenkov, S. Chang, M. H. Fang, B. Qian, E. K. Vehstedt, J. H. Yang, H. M. Pham, L. Spinu and Z. Q. Mao, Phys. Rev. Lett., 2009, 102, 247001 CrossRef
- E. E. Rodriguez, P. Zavalij, P. Y. Hsieh and M. A. Green, J. Am. Chem. Soc., 2010, 132, 10006–10008 CrossRef CAS
- B. C. Sales, A. S. Sefat, M. A. McGuire, R. Y. Jin, D. Mandrus and Y. Mozharivskyj, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 094521 CrossRef
- P. Zajdel, P.-Y. Hsieh, E. E. Rodriguez, N. P. Butch, J. D. Magil, J. Paglione, P. Zavalij, M. R. Suchomel and M. A. Green, J. Am. Chem. Soc., 2010, 132, 13000 CrossRef CAS
- T. J. Liu, X. Ke, B. Qian, J. Hu, D. Fobes, E. K. Vehstedt, H. Pham, J. H. Yang, M. H. Fang, L. Spinu, P. Schiffer, Y. Liu and Z. Q. Mao, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 174509 CrossRef
- R. Viennois, E. Giannini, D. van der Marel and R. Cerny, J. Solid State Chem., 2010, 183, 769–775 CrossRef CAS
- M. Bendele, P. Babkevich, S. Katrych, S. N. Gvasaliya, E. Pomjakushina, K. Conder, B. Roessli, A. T. Boothroyd, R. Khasanov and H. Keller, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 212504 CrossRef
- T. J. Liu, J. Hu, B. Qian, D. Fobes, Z. Q. Mao, W. Bao, M. Reehuis, S. A. J. Kimber, K. Prokes, S. Matas, D. N. Argyriou, A. Hiess, A. Rotaru, H. Pham, L. Spinu, Y. Qiu, V. Thampy, A. T. Savici, J. A. Rodriguez and C. Broholm, Nature Mater., 2010, 31, 716 Search PubMed
- M. D. Lumsden, A. D. Christianson, E. A. Goremychkin, S. E. Nagler, H. A. Mook, M. B. Stone, D. L. Abernathy, T. Guidi, G. J. MacDougall, C. de la Cruz, A. S. Sefat, M. A. McGuire, B. C. Sales and D. Mandrus, Nat. Phys., 2010, 6, 182–186 CrossRef CAS
- J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef CAS
- S. L. Bud'ko, P. C. Canfield, A. S. Sefat, B. C. Sales, M. A. McGuire and D. Mandrus, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 134523 CrossRef
- A. Martinelli, A. Palenzona, M. Tropeano, C. Ferdeghini, M. Putti, M. R. Cimberle, T. D. Nguyen, M. Affronte and C. Ritter, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 094115 CrossRef
- N. C. Gresty, Y. Takabayashi, A. Y. Ganin, M. T. McDonald, J. B. Claridge, D. Giap, Y. Mizuguchi, Y. Takano, T. Kagayama, Y. Ohishi, M. Takata, M. J. Rosseinsky, S. Margadonna and K. Prassides, J. Am. Chem. Soc., 2009, 131, 16944–16952 CrossRef CAS
- H. A. Mook, M. D. Lumsden, A. D. Christianson, S. E. Nagler, B. C. Sales, R. Y. Jin, M. A. McGuire, A. S. Sefat, D. Mandrus, T. Egami and C. dela Cruz, Phys. Rev. Lett., 2010, 104, 187002 CrossRef CAS
- Y. M. Qiu, W. Bao, Y. Zhao, C. Broholm, V. Stanev, Z. Tesanovic, Y. C. Gasparovic, S. Chang, J. Hu, B. Qian, M. H. Fang and Z. Q. Mao, Phys. Rev. Lett., 2009, 103, 067008 CrossRef
- A. D. Christianson, E. A. Goremychkin, R. Osborn, S. Rosenkranz, M. D. Lumsden, C. D. Malliakas, I. Todorov, H. Claus, D. Y. Chung, M. G. Kanatzidis, R. Bewley and T. Guidi, Nature, 2008, 456, 930 CrossRef CAS
- C. Stock, C. Broholm, J. Hudis, H. J. Kang and C. Petrovic, Phys. Rev. Lett., 2008, 100, 087001 CrossRef CAS
- O. Stockert, J. Arndt, A. Schneidewind, H. Schneider, H. S. Jeevan, C. Geibel, F. Steglich and M. Loewenhaupt, Phys. B, 2008, 403, 973 CrossRef CAS
- C. Stock, W. J. L. Buyers, R. Liang, D. Peets, Z. Tun, D. Bonn, D. N. Hardy and R. J. Birgeneau, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 014502 CrossRef
- L. Zhang, D. J. Singh and M. H. Du, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 012506 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: S1-3 contains typical diffraction patterns obtained using the Ge311 and Cu311 monochromator along with more information of the isotropic Gaussian strain. S4 is a contour plot of the temperature variation of the (200) and (111) reflections. Table S1 contains The structural parameters as a function of temperature including goodness-of-fit factors. See DOI: 10.1039/c1sc00114k |
| This journal is © The Royal Society of Chemistry 2011 |