DOI:
10.1039/C1SC00300C
(Edge Article)
Chem. Sci., 2011,
2, 1850-1859
A new polymerisation route to conjugated polymers: regio- and stereoselective synthesis of linear and hyperbranched poly(arylene chlorovinylene)s by decarbonylative polyaddition of aroyl chlorides and alkynes†
Received
17th May 2011
, Accepted 23rd June 2011
First published on 13th July 2011
Abstract
We present here a new polymerisation route for the synthesis of new conjugated polymers. Decarbonylative polyadditions of diyne monomers [bis(4-ethynylphenyl)dimethylsilane, 4,4′-diethynylbiphenyl, and 1,2-bis(4-ethynylphenyl)-1,2-diphenylethene] with respective terephthaloyl dichloride and benzene-1,3,5-tricarbonyl trichloride catalyzed by [Rh(cod)Cl]2/PPh3, [Rh(nbd)Cl]2/PPh3, [RhCp*Cl2]2/PPh3, [Rh(cod)(PPh3)2]+PF6−, or [Rh(cod)(PPh2)(CH2)4(PPh2)]+BF4− (cod = 1,5-cyclooctadiene, nbd = 2,5-norbornadiene, Cp* = pentamethyl cyclopentadienyl), proceed smoothly, producing linear and hyperbranched poly(arylene chloroZ-vinylene)s (PACVs) in regio- and stereoselective manners with high molecular weights (absolute Mw up to 5.31 × 105) in high yields (up to 92%). Model reactions are designed to confirm the chemical structures of the PACVs. The resultant polymers are processable and enjoy high thermal stability. The linear PAVCs can undergo thermal curing at high temperatures and compared with their hyperbranched counterparts, they are more electronically conjugated due to the para-conjugation effect, as revealed by both experimental and theoretical studies. The light emissions of linear PAVCs with twisted tetraphenylethene units are enhanced by aggregate formation, demonstrating an unusual aggregation-enhanced emission characteristic.
Introduction
Exploration of new monomeric units and development of new polymerisation reactions for the construction of functional macromolecules with novel and/or advanced functional properties is a constant theme in current polymer research. In particular, development of new synthetic routes to conjugated polymers is highly rewarding because it will not only contribute to fundamental polymer science but also meet the increasing demand of organic semiconductor materials for low-cost advanced optoelectronic devices, such as flexible organic light-emitting diodes (OLEDs), photovoltaic cells (OPVs), and organic transistors. In this context, carbon–carbon triple bond-based polymer chemistry has made great contributions in preparing conjugated polymers.1 An acetylene monomer contains a triple bond, with one more π-bond than an olefin monomer. When one of the three bonds is opened, the resultant active species is still electronically unsaturated. Acetylenic molecules can also be coupled together, with their triple bonds kept “intact” to yield products with higher degrees of unsaturation. As a typical example, the archetypal polyacetylene has brought us to the threshold of “plastic-electronics era”.2 Due to the enthusiastic and fruitful efforts of polymer chemists and materials scientists in the past decades, acetylenic monomers have been nurtured into a group of versatile building blocks for the construction of π-conjugated polymers and the acetylenic polymerisations have emerged as useful techniques for the syntheses of advanced speciality polymers with novel molecular structures and unique functional properties. For example, triple bond-mediated metathesis or insertion, coupling, addition, and cyclization have been well established to afford acetylenic polymers with different chain structures and dimensionalities such as poly(aryleneethynylene), polydiacetylene, polyarylene, and poly(1,2,3-triazole) as well as their substituted derivatives (Scheme 1).1
Our groups have particular interests in developing organic alkyne reactions into useful tools for the preparation of functional polymers. Employing monoynes, diynes, and triynes as monomers, we have successfully synthesized a large variety of polyacetylenes, polyarylenes, polydiynes, and polytriazoles with linear and hyperbranched structures and regio- and stereoregularities by metathesis, cyclotrimerization, coupling, and click polymerisations.3 Along this research line, there is a vast amount of room to be explored. However, how to develop an efficient and conveniently used polymerisation technique is a great challenge. A number of issues should be carefully concerned and solved including the choice of an efficient catalyst system, monomer scope and functional group tolerance, optimum reaction conditions, control of molecular weights and regio- and stereostructures, and solubility and processability of the resultant polymers.
With these issues in mind, in this work, we took the challenge and developed the rhodium (Rh)-catalyzed decarbonylative reaction of aroyl chlorides with alkynes (Scheme 1) into a useful tool for the construction of poly(arylene chlorovinylene)s (PACVs) with linear and hyperbranched architectures.4PACV is an analog of poly(phenylenevinylene) (PPV), which is a category of well-known conjugated polymer and has found a spectrum of optoelectronic applications such as OLEDs, OPVs, and organic lasers.5 This route offers the advantages of extremely high regio- and stereoselectivities over conventional methods for making PPVs. Proper matching of monomers with different structures and functionalities can readily furnish functional PACVs with high molecular weights in high yields (Schemes 2–4). The PACVs exhibit good solubility, thermal stability, and unique photophysical properties. To our best knowledge, this is the first example for transition metal-catalyzed regio- and stereoselective synthesis of chlorinated PPVs.
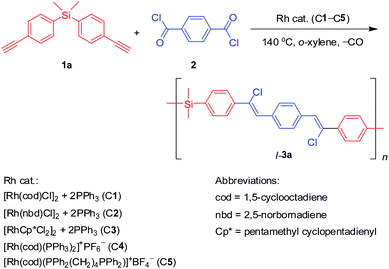 |
| | Scheme 2 Screening of effective catalyst systems. | |
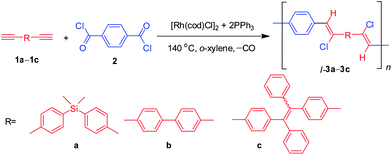 |
| | Scheme 3 Linear polymerisations of diynes with aroyl dichlorides. | |
Results and discussion
Synthesis of linear polymers
Decarbonylative reactions of aroyl chlorides with terminal aromatic alkynes catalyzed by Rh complexes are known to proceed in regio- and stereoselective manners, giving sole Z-vinyl chlorides.4 Due to the simplicity and rich source of these two substrates, we have particular interest in developing this reaction into a useful polymerisation technique for making new conjugated polymers. Through proper design of monomer pairs (A and B) with different functionality, it is envisaged that A2 + B2 and A2 + B3 construction strategies can be readily utilized to synthesize linear and hyperbranched PACVs.
We first performed the screening of catalysts and investigated the catalytic behavior of different rhodium complexes for the polymerisation of model monomers bis(4-ethynylphenyl)dimethylsilane (1a) and terephthaloyl dichloride (2) (Scheme 2). Under the same experimental conditions, binary systems C16,7 and C2,8 and single component of zwitterionic Rh(I) complex C4 can successfully catalyze the polymerisation to give linear PACVs (l-3a) with excellent solubility in common organic solvents. Among them, C1 reveals the best result, giving a polymer with the highest relative weight-average molecular weight (Mw,r 13700) and yield in a reasonable polydispersity index (PDI 2.4) (Table 1, no.1). The C2 and C4 also work well to give polymers with modest molecular weights in good yields (Table 1, nos. 2 and 4). All the polymers are synthesized in regio- and stereoselective manners with sole Z-vinylene structures. On the contrary, only trace amounts of partially soluble product are obtained when the polymerisations are catalyzed by C39 and C5. It is known that aroyl chlorides are capable of reacting with low valent transition-metal species, such as rhodium and palladium species, to produce the aroylchlorometal complexes that can be further transformed into arylchlorometal complexes by decarbonylation at relatively high temperatures.10 Afterwards, the reaction may involve chlororhodation to alkynes to form the arylchlororhodium(III) intermediate followed by reductive elimination of the final product.4 In the catalytic cycle, Rh(I) species is the effective component and repeated transitions between Rh(I) and Rh(III) species are involved. C3 has a stable Rh(III) center, which satisfies the 18-electron rule and thus is not good for the catalysis. Compared with PPh3 in C4, the 1,4-bis(diphenylphosphino)butane ligand in C5 has a higher binding affinity to the rhodium center, which makes it difficult to compromise the ligand association/dissociation balance in the catalytic cycle, thus showing poorer performance.
Table 1
Polymerisations of monomers 1a and 2 in the presence of different Rh complexesa
| No. |
Catalyst
|
Yield (%) |
Sb |
M
w,r
c
|
M
w,r/Mn,rc |
|
Carried out in o-xylene at 140 °C under nitrogen for 8 h in the presence of different rhodium complexes with structures shown in Scheme 2; [1a] = [2] = 0.1 M, [Rh] = 4 mM, [PPh3] = 8 mM.
Solubility (S) tested in common organic solvents such as THF, DCM, and chloroform; √ = completely soluble, Δ = partially soluble.
Estimated by GPC in THF on the basis of a polystyrene calibration; Mw,r = relative weight-average molecular weight; Mn,r = relative number-average molecular weight; Mw,r/Mn,r = polydispersity index.
|
| 1 |
C1 |
81.8 |
√ |
13700 |
2.4 |
| 2 |
C2 |
78.5 |
√ |
12700 |
2.6 |
| 3 |
C3 |
Trace |
Δ |
|
|
| 4 |
C4 |
50.3 |
√ |
5300 |
1.6 |
| 5 |
C5 |
Trace |
Δ |
|
|
Since C1 gives the best polymerisation results, we thus utilized it to study the polymerisation behaviors of other monomer pairs. C1 also works well for the polymerisations of 1b11 and 1c with 2, affording polymers with modest molecular weights in reasonable yields (Scheme 3 and Table 2). Due to a large free volume of the sp3-hybridized silicon, l-3a possesses a good processability. Unlike l-3a, l-3b is insoluble due to its rigid backbone and its facile parallel inter-strand aggregation arising from the strong π–π stacking interactions. Introduction of twisted monomeric units into the backbone may increase the free volume of the polymer chain, which helps reduce the interchain π–π stacking interaction, and hence improves its solubility. Indeed, all-aromatic conjugated polymerl-3c constructed from monomer 1c with twisted, random tetraphenylethene (TPE) (E/Z ratio 45/55) shows good solubility in common organic solvents.
Table 2 Linear polymerisations of diynes with aroyl dichloridesa
| No. |
Polymer
|
[M]o (M) |
[Rh] (mM) |
t (h) |
Yield (%) |
Sb |
M
w,r
c
|
M
w,r/Mn,rc |
|
Carried out in o-xylene at 140 °C under nitrogen in the presence of C1; [PPh3] = 2[[Rh(cod)Cl]2].
Solubility (S) tested in common organic solvents such as THF, DCM, and chloroform; √ = completely soluble, × = insoluble.
Estimated by GPC in THF on the basis of a polystyrene calibration; Mw,r = relative weight-average molecular weight; Mn,r = relative number-average molecular weight; Mw,r/Mn,r = polydispersity index.
|
| 1 |
3a
|
0.1 |
6 |
12 |
91.5 |
√ |
14800 |
2.6 |
| 2 |
3a
|
0.2 |
4 |
6 |
70.7 |
√ |
12100 |
2.9 |
| 3 |
3b
|
0.1 |
4 |
0.3 |
26.3 |
× |
|
|
| 4 |
3c
|
0.08 |
4 |
1 |
61.5 |
√ |
5400 |
2.1 |
| 5 |
3c
|
0.1 |
4 |
1 |
85.6 |
√ |
9300 |
2.8 |
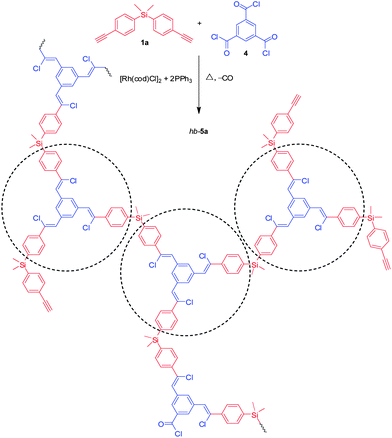
After the successful preparation of linear PACVs, we then utilized this polymerisation technique for the synthesis of hyperbranched polymers. We took an A2 + B3 construction strategy and through careful control of the monomer feed ratio, the polymerisation of silylenephenylenediyne (1a) with benzene-1,3,5-tricarbonyl trichloride (4) conducted in toluene under nitrogen at 110 °C for 10 h proceeds smoothly, producing a hyperbranched PACV (hb-5a) in a yield of 57.1% with an Mw,r value of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Table 3, no. 1). The change of the solvent to o-xylene at an elevated temperature of 140 °C greatly shortens the reaction time to 1 h and generates a polymer in a higher yield (73.4%, Table 3, no. 2). A similar isolation yield was obtained at a higher diyne concentration but the molecular weight of the polymer becomes, however, lower (Table 3, no. 3). Attempts to further improve the reactions by lengthening the time from 1 to 4 h, unfortunately, resulted in the formation of partially soluble product (Table 3, no. 4).
000 (Table 3, no. 1). The change of the solvent to o-xylene at an elevated temperature of 140 °C greatly shortens the reaction time to 1 h and generates a polymer in a higher yield (73.4%, Table 3, no. 2). A similar isolation yield was obtained at a higher diyne concentration but the molecular weight of the polymer becomes, however, lower (Table 3, no. 3). Attempts to further improve the reactions by lengthening the time from 1 to 4 h, unfortunately, resulted in the formation of partially soluble product (Table 3, no. 4).
Table 3 Nonlinear polymerisations of diynes with aroyl trichloridesa
| No. |
Solvent
|
T (°C) |
t (h) |
Yield (wt %)c |
Sd |
M
w,r (Mw,a)e |
M
w,r/Mn,re |
|
Carried out under nitrogen in the presence of C1; [[Rh(cod)Cl]2] = 4 mM, [PPh3] = 8 mM, [1a]/[4] (M M−1) = 0.12/0.08.
[1a]/[4] (M M−1) = 0.16/0.08.
Mass ratio of polymer to monomer.
Solubility (S) tested in common organic solvents such as THF, DCM, and chloroform; √ = completely soluble, Δ = partially soluble.
Estimated by GPC in THF on the basis of a polystyrene calibration; Mw,r = relative weight-average molecular weight; Mw,a = absolute weight-average molecular weight; Mn,r = relative number-average molecular weight; Mw,r/Mn,r = polydispersity index.
|
| 1 |
Toluene
|
110 |
10 |
57.1 |
√ |
14000 |
4.4 |
| 2 |
o-Xylene
|
140 |
1 |
73.4 |
√ |
14100 |
3.3 |
| 3b |
o-Xylene
|
140 |
1 |
71.3 |
√ |
10300 (531000) |
3.0 |
| 4 |
o-Xylene
|
140 |
4 |
85.7 |
Δ |
18300 |
4.2 |
Since most of the polymerisation reactions are conducted at equal mole number of triple bonds and acyl chlorides, they thus have the same probability to remain as terminal groups at the peripheries of the sphere-shaped hyperbranched PACVs after the polymerisation. If one of the functional groups is used in excess, the peripheries of the resultant polymers will be dominated by such functionality. Such strategy can offer the possibility for the post-modification of the polymers by a number of macromolecular engineering methods. In Table 3, no. 3, we increased the mole ratio of triple bonds to acyl chlorides from 1:1 to 4:3 for the polymerisation. The resultant hyperbranched polymer is thus rich in triple bonds at its peripheries, which can be further functionalized by a series of triple bond-based reactions, such as azide–alkyne and thiol–alkyne click reactions. This study is now actively carried out in our group. It is noteworthy to mention that GPC calibrated by linear polystyrene standards can significantly underestimate the molecular weights of hyperbranched polymers due to their globular architecture.12 We thus employed the laser light scattering technique to measure the absolute molecular weight (Mw,a) of hb-5a (Table 3, no. 3). The obtained Mw,a is 531000 (Fig. S1†), which is about 52-fold higher than its Mw,r value.
Model reactions
To confirm the occurrence of the decarbonylative polymerisations and gain insights into the chemical structures of the PACVs, three model reactions were performed as shown in Scheme 5. The model molecules 8 and 9 were further confirmed by single crystal X-ray diffraction technique (See Supporting Information†).
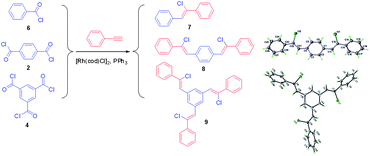 |
| | Scheme 5 Model reactions. | |
Structural characterizations
The polymer products were fully characterized spectroscopically (see Supporting Information for detailed analysis data†). The IR spectrum of l-3a is given in Fig. 1 as an example. The spectra of its monomers 1a and 2 are also shown in the same figure for comparison. The strong absorption bands observed at 3270 cm−1 in 1a and 1725 cm−1 in 2 are associated with their ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H and C
C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively. These two bands disappear after polymerisation. Meanwhile, the absorption band of the aromatic CC skeleton vibration at 1596 cm−1 is intensified. These spectral data indicate that the triple bonds of 1a and acyl chlorides of 2 are consumed and converted to vinylene units by the polymerisation reaction.
O stretching vibrations, respectively. These two bands disappear after polymerisation. Meanwhile, the absorption band of the aromatic CC skeleton vibration at 1596 cm−1 is intensified. These spectral data indicate that the triple bonds of 1a and acyl chlorides of 2 are consumed and converted to vinylene units by the polymerisation reaction.
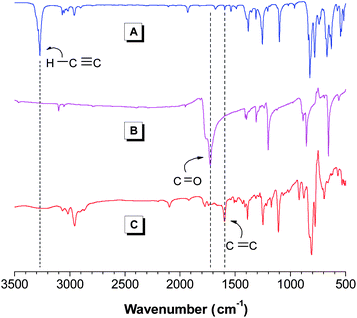 |
| | Fig. 1
IR spectra of monomers (A) 1a and (B) 2 and (C) linear polymerl-3a (sample taken from Table 2, no. 1). | |
Fig. 2 shows the 1H NMR spectra of l-3a, its monomers 1a and 2, and model compound 8. By comparison, the peaks at δ 3.09 and 8.26 in 1a and 2 are assigned to the resonances of their acetylene and benzene protons, respectively, which are absent in the spectrum of l-3a. Instead, a new peak due to the resonance of the Z-vinylene proton emerges at δ 7.08. Compared with the model compound, l-3a shows a much broader absorption in the aromatic region, indicative of its polymeric nature. The 13C NMR spectrum of l-3a displays no resonance peaks of acetylenic carbon atoms of 1a at δ 83.6 and 77.9 and the carbonyl carbon of 2 at δ 167.7, but new resonance peaks in the aromatic carbon region (Fig. 3), which are consistent with those of IR and 1H NMR analyses.
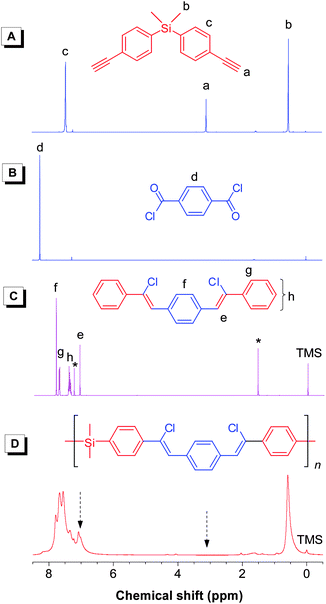 |
| | Fig. 2
1H NMR spectra of monomers (A) 1a and (B) 2, (C) model compound 8, and (D) linear polymerl-3a (sample taken from Table 2, no. 1) in CDCl3 solutions. The peaks for solvent and water are marked with asterisks. | |
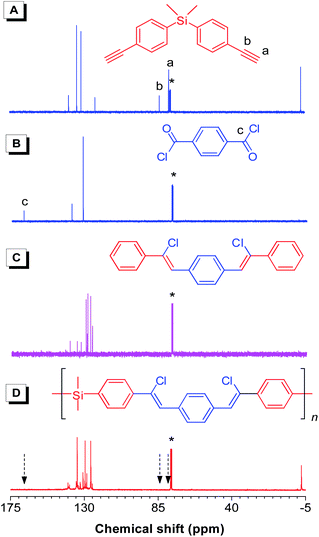 |
| | Fig. 3
13C NMR spectra of monomers (A) 1a and (B) 2, (C) model compound 8, and (D) linear polymerl-3a (sample taken from Table 2, no. 1) in CDCl3 solutions. The solvent peaks are marked with asterisks. | |
As stated before, we changed the monomer pair from 1a/2 to 1a/4, a hyperbranched PACV was obtained. Figs. S2, 4, and S3 show the IR, 1H NMR, and 13C NMR spectra of hb-5a constructed from 1a and 4.† By comparison with the spectra of the monomers and the model compound (9), we can confirm the successful synthesis of a hyperbranched PACV (hb-5a) as shown in Scheme 4.
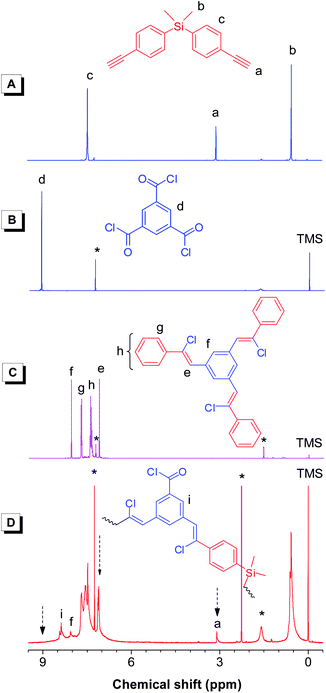 |
| | Fig. 4
1H NMR spectra of monomers (A) 1a and (B) 4, (C) model compound 9, and (D) a hyperbranched polymerhb-5a (sample taken from Table 3, no. 3). The peaks for solvents (CDCl3 and o-xylene) and water are marked with asterisks. | |
Determination of degree of branching
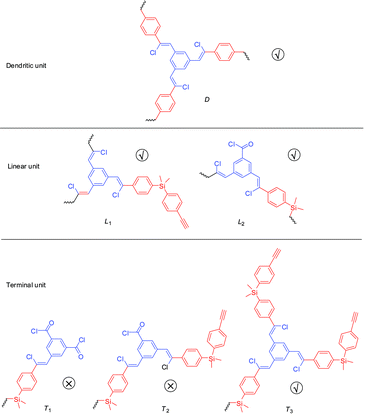
An important parameter for a hyperbranched polymer is its degree of branching (DB),13 which is usually determined by 1H NMR spectral analysis. From the structural viewpoint, if the mutually reactive groups from the two monomers have the same mole number, six possible structural units may exist in an hb-5a: one dendritic unit (D), two linear units [one with an unreacted ethynyl group (L1) and another with an unreacted acyl chloride (L2)], and three terminal units [one with two acyl chloride groups (T1), one with an acyl chloride group and an ethynyl group (T2), and one with two ethynyl groups (T3)]. It is difficult to identify these units and differentiate them from each other by NMR spectroscopy. Thus, these six parameters cannot be calculated directly. If we tune the feed ratio of the two functional groups to make one of them in excess, the distribution of these six basic units will be altered and some of them may be excluded, which may offer the possibility to calculate the DB.
The 1H NMR spectrum of hb-5a prepared by a feed ratio of 2:1 of 1a to 4 (4:3 of mole number of triple bond to acyl chloride unit) in Fig. 4D suggests that two terminal units T1 and T2 can be excluded due to the end capping by excess triple bonds. Based on the assumption that no loop is formed during the polymerisation, DB of a hyperbranched polymer is defined as the ratio of the number of its dendritic and terminal units to its total structural units.14 According to this definition, DB of hb-5a can be expressed as follows:
| | 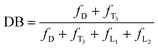 | (1) |
where
fD,
fL1,
fL2, and
fT3 are the molar fraction of the specific basic structural units mentioned above (
Chart 1). By analyzing the NMR spectra of monomers, model compound, and the resultant
polymer (
Fig. 4), the following relationships are established:
| | 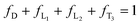 | (2) |
| | 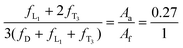 | (3) |
| | 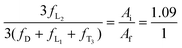 | (4) |
where
Aa,
Af, and
Ai are the integrated areas of the resonance peaks a, f, and i as labeled in
Fig. 4D. In addition, the well-known and generally accepted “principle of equal reactivity of functional groups” is applied to our system.
15 Thus, according to
Chart 1,
eqn (5) holds:
Combining eqn (2)–(5), we obtain the values of molar fractions of the basic structural units
Eventually, the DB value of hb-5a is determined to be 0.30 according to eqn (1). This value is lower than those of the “conventional” hyperbranched polymers with the most probable value of ∼0.513,16 due to two reasons. One is that this polymer is not prepared under a stoichiometric balance of the two mutually reactive groups, and another factor is due to the structural crowdedness of monomer 4, which makes the reaction of remaining acyl chloride moiety difficult after the first two are consumed. As a result, this renders the formation of a large fraction of linear units (fL2) and hence lowers the DB of the polymer. To achieve polymers with high DB values, it is better to select monomers with more spatially separated functional groups. Currently, we are expanding this research in the hope of making hyperbranched conjugated polymers with different chromophoric units and controllable DB values.
Thermal properties
The thermal properties of the PACVs are evaluated by thermogravimetric analysis (TGA). As shown in Fig. 5, both l-3a and hb-3a enjoy outstanding thermal stability, with their degradation temperatures for 5% weight loss (Td's) occurring at ∼350 °C. l-3a carbonizes in a higher yield (up to 65%) than hb-5a (∼50%) when pyrolyzed at 800 °C. To understand the difference, we studied their thermal transitions by differential scanning calorimetry (DSC). The thermogram of l-3a recorded during the first heating scan displays an exothermic peak at 268 °C. Since l-3a does not lose its weight below 350 °C, this peak is thus not due to the chain scission but associated with the thermally induced cross-linking of vinyl chloride groups. The second heating scan gives an even line in the same temperature region, suggesting the completion of the thermal crosslinking reaction after the first heating cycle. On the contrary, no obvious thermal transitions are detected in hb-5a in both first and second heating cycles. This result indicates that the hyperbranched polymer has not undergone the crosslinking reaction in this temperature region, probably due to its spatially branched structures, which have lowered the probability for the vinyl chloride groups to approach each other. Consequently, the thermal resistance is lowered at high temperatures. The involved cross-linking mechanism is actively underway in our group.
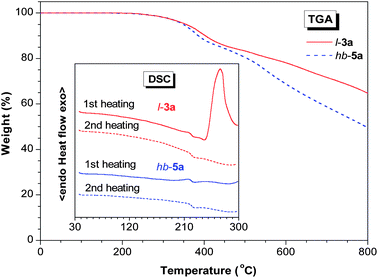 |
| | Fig. 5
TGA and DSC thermograms of l-3a and hb-5a (samples taken from sample taken from Table 2, no.1 and Table 3, no. 3, respectively) recorded under nitrogen at a heating rate of 10 °C min−1. | |
Photophysical properties
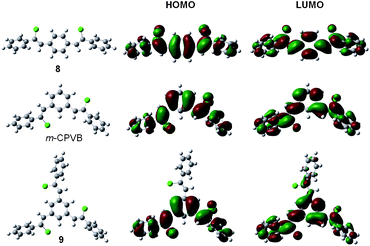
Fig. 6A shows the UV absorption spectra of model compounds 8 and 9, and polymersl-3a and hb-5a in THF solutions. The absorption spectrum of 8 is peaked at 330 nm, while the absorption maximum of 9 is located at 300 nm. This is in some sense not surprising because for compounds with chromophore-conjugated bridge-chromophore structures, the ones with para-bridges show better electronic communications and hence smaller band gaps than those with meta-linkers due to electronic decoupling of the chromophores in the ground electronic state.17 As 8 and 9 are para- and meta-substituted, respectively, this explains why the absorption of the former is ∼30 nm red-shifted from that of the latter. Polymersl-3a and hb-5a contain 8 and 9 as basic structural units, respectively, and their absorption maxima are also observed at ∼330 and 300 nm. Their profiles are, however, much broader, indicative of more conjugated electronic structures arising from the potential σ–π conjugation between the silylene and π-conjugation units in the backbones.18 Compared with the solutions, polymer thin films absorb more broadly, suggestive of the possibility of interchain interactions (Fig. S4†). To prove the meta-conjugation effect, we carried out theoretical calculation of 8 and 9 based on the density functional theory at the B3LYP/6-31G(d) level.19 Meanwhile, the frontier orbital pictures of (Z,Z)-1,4-bis(2-chloro-2-phenylvinyl)benzene (m-CPVB), one type of basic structural units of hb-5a, was also calculated for comparison. The highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO) and their energy values are shown in Fig. 7 and Table 4. It can be seen that one branch of 9 shows no contribution to its HOMO and LUMO and the band gap of 8 (∼3.63 eV) is much narrower than those of 9 (∼4.21 eV) and m-CPVB (∼4.18 eV), suggestive of a better conjugation. The experimental HOMO/LUMO energy levels of 8 and 9 determined by cyclic voltammetry show a similar trend to that of DFT study (Fig. S5†). No apparent redox peaks were observed for l-3a and hb-5a in the designed scanning window. The theoretical study is consistent with the experimental result and both of them verify the meta-conjugation effect.
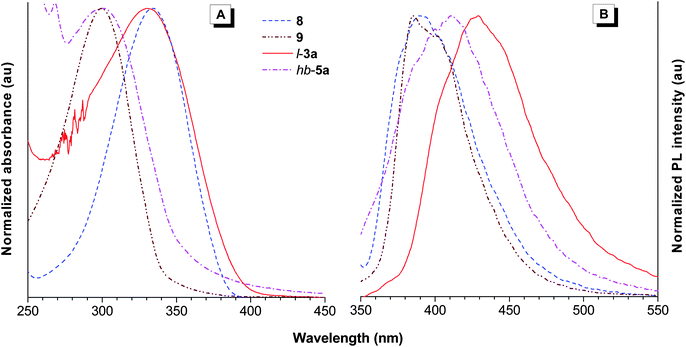 |
| | Fig. 6 Normalized (A) UV and (B) PL spectra of model compounds 8 and 9, l-3a (sample taken from Table 2, no. 1), and hb-5a (sample taken from Table 3, no. 3) in THF solutions. Concentrations: 10−5 M; excitation wavelength: 330 nm (8 and l-3a) and 300 nm (9 and hb-5a). | |
Table 4 Energy levels of HOMO and LUMO of 8, m-CPVB, and 9
| |
8
|
m-CPVB
|
9
|
|
The values in parentheses are experimentally derived from the cyclic voltammetry measurement.
Calculated from the absorption onset.
E
LUMO = EHOMO + EGap.
|
| HOMO (eV)a |
−5.53 (−5.88) |
−5.76 |
−5.81 (−5.98) |
| LUMO (eV)c |
−1.90 (−2.63) |
−1.58 |
−1.60 (−2.34) |
| Gap (eV)b |
3.63 (3.25) |
4.18 |
4.21 (3.64) |
Upon photoexcitation, the THF solutions of l-3a and hb-5a give photoluminescence (PL) at 430 and 411 nm, which are red-shifted from those model compounds 8 (391 nm) and 9 (385 nm) by 39 and 26 nm, respectively, further corroborating the meta-conjugation and σ–π conjugation effects. Introduction of heavy atoms such as I, Br, and Cl, into the polymers may enhance the possibility of phosphorescence because intersystem crossing process is favored.20 It is known that phosphorescence of organic luminophores is rarely observed at ambient conditions and the energy of the lowest vibrational level of the triplet state is lower than that of the singlet state. We thus measured the PL of 8, 9, l-3a, and hb-5a at low temperature to check whether new emission peaks would appear at longer wavelengths (Fig. 8). In the frozen state by liquid nitrogen, the emissions of the models and polymers are greatly enhanced due to the reduced thermal agitation through collision interaction with solvent molecules and the restriction of intramolecular molecular motions (vibration and rotation). No redder emission peaks appears and only some fine structures corresponding to specific fluorescence energy bands are discerned. Consequently, these facts rule out the possibility of phosphorescence.
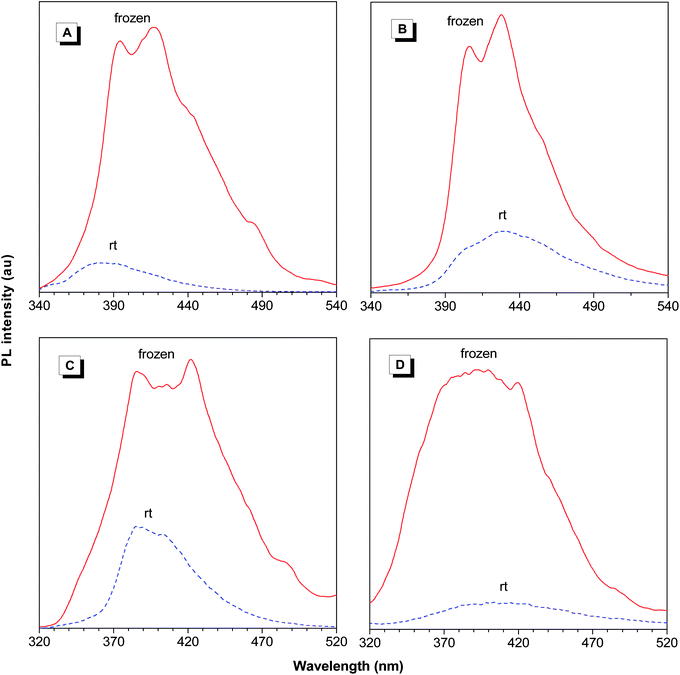 |
| | Fig. 8 Temperature-dependent PL spectra of (A) model compound 8, (B) l-3a, (C) model compound 9, and (D) hb-5a in THF solutions. Concentrations: 10−5 M. Excitation wavelength: 330 nm (8 and l-3a) and 300 nm (9 and hb-5a). | |
Our group is interested in studying a novel class of luminogens with aggregation-induced emission (AIE) characteristics: molecules are non-emissive in dilute solutions but induced to emit intensively by aggregate formation.21,22 The AIE phenomenon is opposite to the notorious aggregation-caused quenching (ACQ) effect. In the condensed phase, the luminogenic molecules are located in the immediate vicinity, which promotes the formation of such detrimental species as excimers and exciplexes and leads to undesired nonradiative transitions.23 The ACQ effect reduces the performance of light-emitting devices like OLEDs. Development of AIE materials is both of academic and practical values. Academically, the photophysical knowledge will be enriched and new theory regarding how to design efficient luminogenic materials in the aggregate state will be spawned. Practically, luminogenic materials with inherently high emission efficiency and spectral stability in the solid state can be obtained and thus no painstaking efforts are needed to artificially hamper the natural process of luminophore aggregation in the fabrication of optoelectronic devices. Current studies mainly focus on small AIE molecules with twisted structures. To overcome the processing disadvantages of low molecular weight compounds, we expand our AIE research program from low molecular weight to polymeric systems.24
We utilized 1,2-bis(4-ethynylphenyl)-1,2-diphenylethene (1c) as one of the building blocks for the construction of linear PACVl-3c and studied its optical properties. The monomer absorption maximum of l-3c is found at ∼370 nm, which is red-shifted by 40 nm from that of 8 due to the conjugation between 8 and the TPE unit. Upon photoexcitation, l-3c emits a green light at 520 nm. Addition of 90 vol% of water (fw = 90%), a nonsolvent for the polymer, into its THF solution has enhanced its light emission, while keeping the spectral profile unchanged, showing an aggregation-enhanced emission (AEE) behavior (Fig. 9B). On the contrary, the emission of 8 was weakened, red-shifted, and broadened under the same conditions, indicative of formation of excimers. Our group has proposed that restriction of intramolecular motion (RIM) is the main cause for the AIE effect.22 In the solution state, the intramolecular motions are active, which serve as nonradiative pathways for the excitons to decay. In the aggregate state, such motions are restricted, which block the relaxation channels and populate the radiative decay, thus turning on the light emission of the luminophores. The TPE units are linked together by covalent bonds, which have partially restricted their motions and hence made the polymer somewhat emissive in THF solution. In THF/water mixture, the RIM process is further strengthened, which has resulted in enhanced emission. It is important to mention here that traditional conjugated polymers suffer from the interstrand parallel aggregation and thus show decreased emission efficiency and spectral instability. This result shown here provides a good example of how to build highly emissive polymers with good spectral stability in the aggregate state.
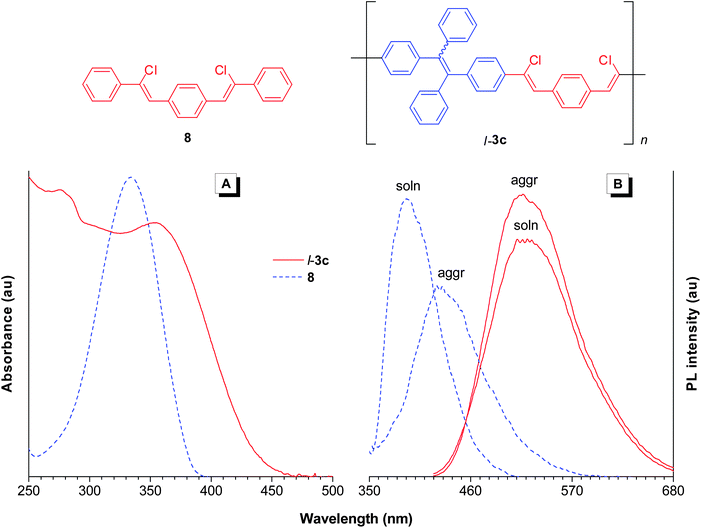 |
| | Fig. 9 (A) UV spectra of model compound 8 and l-3c in THF solutions. (B) Their PL spectra in THF solutions (soln) and THF/water (1/9 v/v) mixtures (aggr). Concentrations: 10−5 M. Excitation wavelength (nm): 330 (8) and 355 (l-3c). | |
Conclusions
In this work, we have developed a new polymerisation route for the synthesis of new electronically conjugated polymers. The Rh-catalyzed decarboxylative polyadditions of aroyl chlorides and alkynes are performed in regio- and stereoselective manners, producing sole Z-PACVs with high molecular weights in high yields. Utilizing the A2 + B2 and A2 + B3 strategies, PACVs with linear and hyperbranched architectures are facilely constructed and thoughtfully characterized by standard spectroscopic techniques. Rational model reactions are performed to help elucidate the chemical structures of the polymers. The determination of DB of the hyperbranched PACV (hb-5a) is demonstrated. The absolute molecular weight of hb-5a determined by LLS is much higher than its relative one estimated by GPC, substantiating its branched structure. The polymers enjoy good solubility and thermal stability. Thermal curing occurs in linear PACVs in our monitored temperature window. The photophysical properties of the model compounds and their polymers are studied with the assistance of theoretical calculation and the meta-conjugation effect is discussed. Polymerl-3c shows an unusual AEE characteristic and its construction strategy inspires us how to build conjugated polymers with high emission efficiency in the aggregate state.
Acknowledgements
The work described in this paper was partially supported by the Research Grants Council of Hong Kong (603509 and HKUST2/CRF/10), the Innovation and Technology Commission (ITP/008/09NP), the University Grants Committee of Hong Kong (AoE/P-03/08), and the National Science Foundation of China (20974028). B.Z.T. thanks the support from Cao Gaungbiao Foundation of Zhejiang University.
Notes and references
-
(a) J. Z. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799 CrossRef CAS;
(b) B. Z. Tang, Macromol. Chem. Phys., 2008, 209, 1303 CrossRef CAS.
-
(a) H. Shirakara, Angew. Chem. Int. Ed., 2001, 40, 2575 Search PubMed;
(b) A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581 CrossRef CAS;
(c) A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591 CrossRef CAS.
-
(a) A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522 RSC;
(b) M. Häußler and B. Z. Tang, Adv. Polym. Sci., 2007, 209, 1 CrossRef;
(c) J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745 CrossRef CAS.
- K. Kokubo, K. Matsumasa, M. Miura and M. Nomura, J. Org. Chem., 1996, 61, 6941 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS.
- B. Z. Tang, W. H. Poon, S. M. Leung, W. H. Leung and H. Peng, Macromolecules, 1997, 30, 2209 CrossRef CAS and references therein.
-
(a) E. Jellema, P. H. M. Budzelaar, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2007, 129, 11631 CrossRef CAS;
(b) E. Jellema, A. L. Jongerius, A. J. C. Walters, J. M. M. Smits, Joost N. H. Reek and B. de Bruin, Organometallics, 2010, 29, 2823 CrossRef CAS.
- C. White, A. Yates and P. M. Maitlis, Inorg. Synth., 1992, 29, 228 CrossRef CAS.
- J. W. Kang, K. Moseley and P. M. Maitlis, J. Am. Chem. Soc., 1969, 91, 5970 CrossRef CAS.
-
(a)
J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organotransition Metal Chemistry; University Science Books: Mill Valley, 1987 Search PubMed;
(b)
H. M. Colquhoun, D. J. Thompson and M. V. Twigg, Carbonylation; Plenum Press: New York, 1991 Search PubMed.
- H. Peng, L. Cheng, J. Luo, K. Xu, Q. Sun, Y. Dong, F. Salhi, P. P. S. Lee, J. Chen and B. Z. Tang, Macromolecules, 2002, 35, 5349 CrossRef CAS.
-
(a) K. E. Uhrich, C. J. Hawker, J. M. J. Frechet and S. R. Turner, Macromolecules, 1992, 25, 4583 CrossRef CAS;
(b) Z. Muchtar, M. Schappacher and A. Deffieux, Macromolecules, 2001, 34, 7595 CrossRef CAS;
(c) S. M. Grayson and J. M. J. Frechet, Macromolecules, 2001, 34, 6542 CrossRef CAS;
(d) J. Liu, L. Zhang, J. W. Y. Lam, C. K. W. Jim, Y. Yue, R. Deng, Y. Hong, A. Qin, H. H. Y. Sung, I. D. Williams, G. C. Jia and B. Z. Tang, Macromolecules, 2009, 42, 7367 CrossRef CAS.
-
(a) B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924 CrossRef CAS;
(b) C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS;
(c) Y. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS.
-
(a) C. J. Hawker, R. Lee and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4583 CrossRef CAS;
(b) H. Frey and D. Holter, Acta Polym., 1999, 50, 67 CrossRef CAS.
-
R. J. Young and P. A. Lovell, Introduction to Polymers, 2nd ed.; Chapman & Hall: London, 1991 Search PubMed.
-
(a) D. Y. Yan, A. H. E. Muller and K. Matyjaszewski, Macromolecules, 1997, 30, 7024 CrossRef CAS;
(b) K. Ishizu, K. Tsubaki, A. Mori and S. Uchida, Prog. Polym. Sci., 2003, 28, 27 CrossRef CAS;
(c) D. Grebel-Koehler, D. J. Liu, S. De Feyter, V. Enkelmann, T. Weil, C. Engels, C. Samyn, K. Müllen and F. C. De Schryver, Macromolecules, 2003, 36, 578 CrossRef CAS.
-
(a) S. Karabunarliev, M. Baumgarten, N. Tyutyulkov and K. Müllen, J. Phys. Chem., 1994, 98, 11892 CrossRef CAS;
(b) M. Tammer, L. Horsburgh, A. P. Monkman, W. Brown and H. D. Burrows, Adv. Funct. Mater., 2002, 12, 447 CrossRef CAS;
(c) Q. Chu, Y. Pang, L. Ding and F. E. Karasz, Macromolecules, 2002, 35, 7569 CrossRef CAS;
(d) M. M. Oliva, J. Casado, G. Hennrich and J. T. L. Navarrete, J. Phys. Chem. B, 2006, 110, 19198 CrossRef;
(e) S. Y. Hong, D. Y. Kim, C. Y. Kim and R. Hoffmann, Macromolecules, 2001, 34, 6474 CrossRef CAS;
(f) A. Pogantsch, A. K. Mahler, G. Hayn, R. Saf, F. Stelzer, E. J. W. List, J.-L. Bredas and E. Zojer, Chem. Phys., 2004, 297, 143 CrossRef CAS;
(g) J.-S. Yang, Y.-R. Lee, J.-L. Yan and M.-C. Lu, Org. Lett., 2006, 8, 5813 CrossRef CAS.
-
(a) J. Liu, R. Zheng, Y. Tang, M. Häussler, J. W. Y. Lam, A. Qin, M. Ye, Y. Hong, P. Gao and B. Z. Tang, Macromolecules, 2007, 40, 7473 CrossRef CAS;
(b) J. C. Sanchez and W. C. Trogler, Macromol. Chem. Phys., 2008, 209, 1527 CrossRef CAS;
(c) G. Kwak, A. Takagi and M. Fujiki, Macromol. Rapid Commun., 2006, 27, 1561 CrossRef CAS;
(d) S. Yamaguchi and K. Tamao, J. Chem. Soc., Dalton Trans., 1998, 3693 RSC.
-
M. J. Frisch, et al., Gaussian 03, rev B.01; Gaussian, Inc.: Pittsburgh, PA, 2003 Search PubMed.
-
B. Valeur, Molecular Fluorescence: Principle and Applications; Wiley-VCH: Weinheim, Germany, 2002 Search PubMed.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- For reviews, see:
(a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC;
(b) J. Liu, J. W. Y. Lam and B. Z. Tang, J. Inorg. Organomet. Polym. Mater., 2009, 19, 249 CrossRef CAS.
-
J. B. Birks, Photophysics of Aromatic Molecules; Wiley: London, 1970 Search PubMed.
-
(a) J. Liu, Y. Zhong, J. W. Y. Lam, P. Lu, Y. Hong, Y. Yu, Y. Yue, M. Faisal, H. H. Y. Sung, I. D. Williams, K. S. Wong and B. Z. Tang, Macromolecules, 2010, 43, 4921 CrossRef CAS;
(b) K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 1418 CrossRef CAS;
(c) J. Liu, Y. Zhong, P. Lu, Y. Hong, J. W. Y. Lam, M. Faisal, Y. Yu, K. S. Wong and B. Z. Tang, Polym. Chem., 2010, 1, 426 RSC;
(d) A. J. Qin, C. K. W. Jim, Y. H. Tang, J. W. Y. Lam, J. Liu, F. Mahtab, P. Gao and B. Z. Tang, J. Phys. Chem. B, 2008, 112, 9281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section for general information, synthesis of monomers, polymers, and model compounds, and detailed characterization data of monomer 1c, model compounds, and polymers. Crystal data for 8 and 9 (Tables S1 and S2), scattering vector dependence of Rayleigh ratio [KC/Rvv(q)] of hb-5a in THF (Fig. S1), IR spectra of 1a, 4, and hb-5a (Fig. S2), 13C NMR spectra of 1a, 4, and hb-5a (Fig. S3), normalized absorption spectra of polymer thin films (Fig. S4), cyclic voltammograms of 8, 9, l-3a, and hb-5a (Fig. S5). CCDC reference numbers 826225 and 826226. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00300c |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.