Synthesis of organocatalysts using non-covalent chemistry; understanding the reactivity of ProNap, an enamine-type organocatalyst that can self assemble with complementary co-catalysts†
José A.
Fuentes
,
Tomas
Lebl
,
Alexandra M. Z.
Slawin
and
Matthew L.
Clarke
*
School of Chemistry, University of St Andrews, EaStCHEM, St Andrews, Fife, UK. E-mail: mc28@st-andrews.ac.uk; Fax: +(44) 1334 463808; Tel: +(44) 1334 463850
First published on 26th July 2011
Abstract
A promising but relatively unexplored approach to tuning asymmetric catalysts is to design catalysts that can self-assemble with a family of complementary co-catalysts. In order to study the mechanism of self-assembled enamine catalysts that are tagged with a supramolecular receptor, a new series of more soluble complementary co-catalysts have been developed. The formation of hydrogen-bonded supramolecular pre-catalysts was established by NMR and X-ray crystallography. NMR studies have identified an enamine intermediate in the reactions between the supramolecular organocatalyst ProNap and aldehydes in which the co-catalysts are hydrogen-bonded. The formation of imidazolidinones from ketones is a reversible process that is likely to coincide with formation of trace amounts of the productive enamine intermediate. The equilibrium is reached much faster in the presence of the complementary co-catalysts, explaining the faster initial rates and greater productivity when ketones are used in stoichiometric quantities relative to the Michael acceptor in asymmetric Michael reactions. These imidazolidinones are a parasitic dead-end in the reaction of aldehydes, both for the self-assembled Proline derivatives and for simple Proline-derived amides. Catalysts prepared from (S)-pyrrolidine-3-carboxylic acid are immune to this and high-yielding nitro-Michael reaction using aldehydes can be achieved.
Introduction
Bifunctional catalysts have fascinated chemists for many years. The majority of bifunctional catalysts are able to simultaneously bind two different substrate molecules and thereby activate them towards a reaction.1 A relatively unexplored but emerging approach is to design a catalyst that can simultaneously bind a substrate and a co-catalyst. Such approaches might enable access to a catalyst structure or function that is not readily accessed using normal catalyst synthesis, facilitate high-throughput optimisation, enable the installation of extra functions into a catalyst, or the switching of the properties of the catalyst. However, these possibilities are just that; possibilities that may arise once a greater understanding of the validity of such approaches is available. The majority of studies in supramolecular catalysis have concentrated on adding a second ligand to a transition metal catalyst or binding of secondary metals and anions.2 Our contrasting interest in this area has been in the use of complementary hydrogen-bonding interactions to modify intact catalysts in a well-defined manner.3 In 2007, we communicated some of our initial findings using ‘ProNap’, 1, a Proline analogue that is tagged with amido naphthyridine receptors, and therefore designed to operate as an enamine catalyst, and simultaneously bind complementary co-catalysts in the form of a range of substituted pyridinones (Fig. 1).3a These results demonstrated that the properties of a functioning chiral catalyst could be radically altered by supramolecular modification. In the arena of organocatalysis, several related approaches to self-assembled catalysts have since appeared, perhaps indicating a significant future for these types of catalysts.4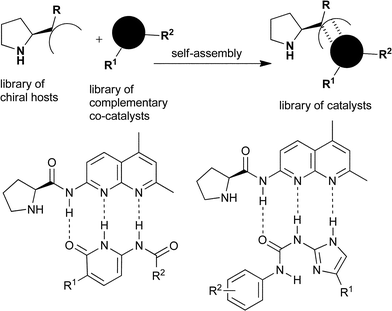 | ||
| Fig. 1 A representation of the general concept of modifying intact catalysts by self-assembly, and the two systems under investigation here. | ||
Our initial communication demonstrated this unusual phenomenon,3a but did not address the mechanism of action of these relatively complex systems. In our view, this is quite an important issue for the development and streamlining of this type of approach, and we therefore commenced a project aimed at identifying the intermediates in these reactions. There were several objectives and questions to be answered that we felt would be enabling to further studies by our group and others. These were: (i) to identify the structure of the self-assembled catalysts formed from ProNap 1 and the co-catalysts, (ii) to understand why these catalysts give such poor results when aldehydes were used as substrates, (iii) to explain why the positive attributes of the complementary co-catalysts were cancelled out by the addition of DMSO, (iv) to shed light on the faster initial rates observed when the co-catalysts were present, and their ability to function without excess ketone, and finally (v) to identify the key enamine intermediate and investigate if the co-catalyst is hydrogen bonded at this stage. Given the nature of enamine catalysis and the solubility of strongly hydrogen bonded molecules, this turned out to be rather challenging, requiring the development of a new library of co-catalysts. In this article we report studies that shed light on all these questions.
Results and discussion
A few of the more informative results from our first studies with ProNap 1 along with some of our first follow-up experiments are shown in Table 1. Given that H-bond donors have since emerged as catalysts for the nitro-Michael addition,5 we investigated some non-complementary co-catalysts in combination with ProNap 1. The striking enhancement of enantioselectivity only occurs with the complementary co-catalysts, 3 to 5 (Fig. 2 and Table 1, entries 3–5). Thio-urea 8 was chosen since it would be at least as good a receptor for the nitro-alkene as the pyridinones,5d,t and therefore to explore if this alternative role of the co-catalyst was more important. However, the use of 8 as co-catalyst has essentially no effect on the enantioselectivity.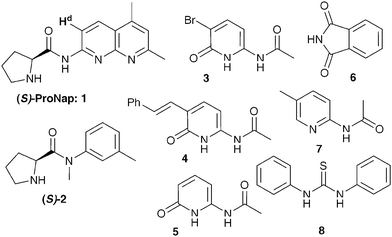 | ||
| Fig. 2 Selected organocatalysts and complementary co-catalysts. | ||
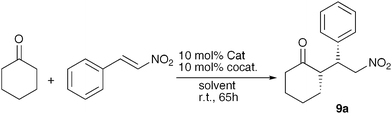
| Entry | Cat. | Cocat. | Solvent | C[%]a | dra | ee[%]b |
|---|---|---|---|---|---|---|
| a Determined by 1H NMR. Reactions were run using 10 mol % catalyst, 4.8 equivalents of ketone at rt for 65 h unless indicated. b Determined by HPLC using Chiralpak AD column. c 19.2 equiv. of ketone, 91 h. | ||||||
| 1c | (S)-Pro | none | CHCl3 | 1 | n.d. | 27 |
| 2c | (S)-1 | none | CHCl3 | 87 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
15 |
| 3 | (S)-1 | 3 | CHCl3 | 82 | 31:1 |
47 |
| 4 | (S)-1 | 4 | CHCl3 | 91 | 42:1 |
32 |
| 5 | (S)-1 | 5 | CHCl3 | 98 | 59:1 |
72 |
| 6 | (S)-2 | none | CHCl3 | 93 | 20:1 |
35 |
| 7 | (S)-2 | 5 | CHCl3 | 95 | 28:1 |
31 |
| 8 | (S)-1 | AcOH | CHCl3 | 72 | 28:1 |
23 |
| 9 | (S)-1 | 6 | CHCl3 | 43 (41 h) | 17:1 |
8 |
| 10 | (S)-1 | 7 | CHCl3 | 43 (70 h) | 20:1 |
14 |
| 11 | (S)-1 | 8 | CHCl3 | 92 (72 h) | 18:1 |
10 |
| 12 | (S)-1 | 5 | EtOH | 12 | 15:1 |
22 |
| 13 | (S)-1 | 5 | DMSO | traces | n.d. | n.d. |
In addition, DMSO or EtOH, solvents known to disrupt hydrogen bonding, completely diminish the positive co-catalyst effect (entries 12 and 13). This is consistent with, but does not prove, a self-assembly process between ProNap 1 and the complementary co-catalysts. In addition, we prepared a non-tagged Proline amide catalyst, compound 2 (chosen as a close analogue in shape that can be prepared in one step), lacking complementary hydrogen bonding motifs and found the pyridinone co-catalysts have no effect on Michael reactions catalysed by 2. The above data is reasonably compelling, but the ultimate proof would be the characterisation of the enamine intermediate both in the presence and absence of the complementary co-catalysts. However, any form of mechanistic study instantly fell into problems due to the low solubility of the pyridinone co-catalysts. In order to achieve this, we elected to develop an alternative class of complementary co-catalyst for ProNap 1 that would have greater solubility than the pyridinones.6a
Wilson and co-workers developed ureidoimidazoles 10 and 12 (Fig. 3) to use as complementary partners of an amidoisocytosine displaying an acceptor-acceptor-donor (AAD) array,6b but since they display a similar linear DDA array to pyridinones seemed a highly suited choice to use as co-catalysts with ProNap 1. Of paramount importance for us was the fact that the introduction of the tert-butyl group in the imidazole ring greatly improved the solubility in CDCl3 of the otherwise poorly soluble compounds. Ureidoimidazoles 11 and 13 were prepared using similar procedures (see ESI†) to those previously reported for the synthesis of 10 and 12.
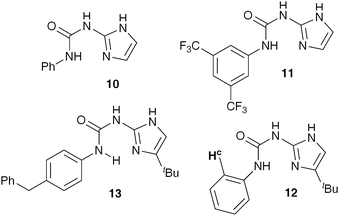 | ||
| Fig. 3 New more soluble co-catalysts used in NMR studies. | ||
Before the catalytic and mechanistic studies with the new co-catalysts were initiated, we turned our attention to investigate the strength of the complementary hydrogen bonding between the host (ProNap 1) and the guest (ureidoimidazole 12), which has been used for most of the mechanistic studies here. The binding constant value was calculated by 1H NMR measurements in CDCl3. Dilutions (1:1 model) were performed at 25 °C from 0.05 M to 0.005 M. The Hc resonance (Fig. 3, compound 12) was selected as a non-acidic proton and because it was used in the work of Wilson.6b It was monitored as it moved downfield with increasing concentration and then analysed using a curve fitting programme to determine Ka (see ESI†). Analysis of the data revealed that the equilibrium between ProNap 1–12 duplex and the free components was 800:1, strong enough for our purposes, since the catalytic reactions were run at 12.5 × 10−3 M catalyst concentration. An X-ray diffraction study on single crystals of self-assembled proNap 1–12 grown from the slow evaporation a chloroform/acetonitrile solution confirms the proposed structure of the host–guest pair as shown in Fig. 4.
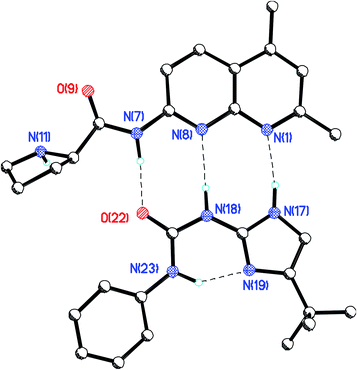 | ||
| Fig. 4 X-ray crystal structure of the catalyst-co-catalyst duplex. | ||
With the new class of more soluble co-catalyst in hand, ureidoimidazoles 10–12 were tested on the asymmetric nitro-Michael reaction between cyclohexanone and nitroalkenes. The behaviour of this new class of co-catalysts is similar to that of the pyridinone family. All the new catalyst combinations have a significant positive effect on both the reaction rate and the stereoselectivity of the reaction. Illustrative examples can be found in Table 2. Low polarity solvents like CH2Cl2, CHCl3 and benzotrifluoride were preferred, but solvents of increased polarity, such as Me-THF can also be used without detriment in the enantioselectivity of the reaction. Ureidoimidazole 10 seems to be the co-catalyst of choice for β-nitrostyrene (entry 10), but for 4-MeO- and 3-NO2- β-nitrostyrene, ureidoimidazole 12 gave the higher enantioselectivity in the reaction (entries 15 and 18). The fact that different substrates optimally prefer different co-catalysts, shows the potential of this approach to optimise enantioselectivity in a reaction without changing the parent catalyst. Now we had a complementary co-catalyst (12) that promotes the activity and enantioselectivity of ProNap 1 and, crucially, has good solubility, we re-initiated our mechanistic studies to identify the key enamine intermediate7,8 and investigate if the co-catalyst is hydrogen bonded at this stage of the reaction.
| Entrya | R1 | Cocat. | Solvent | Conv. [%]b | drc | ee [%]d |
|---|---|---|---|---|---|---|
| a Reactions carried out at rt in 2 ml of dry solvent pre-stirring co-catalyst and pre-catalyst, (R)-1 for 30 min and using 4.8 eq of ketone for 65 h unless indicated. b Michael products/(Michael products + nitroalkene) × 100 determined by 1H-NMR integration. c Ratio determined by 1H-NMR integration after work-up. d Determined by chiral HPLC. e 24 h. f 1 eq. of ketone. g 20 mol% 12. | ||||||
| 1 | Ph | none | CHCl3 | 77 | 29:1 |
11 |
| 2 | Ph | 10 | CHCl3 | 99 | 60:1 |
55 |
| 3 | Ph | 11 | CHCl3 | 97 | 38:1 |
58 |
| 4 | Ph | 12 | CHCl3 | 98 | 48:1 |
47 |
| 5 | Ph | none | PhCF3 | 53 | 28:1 |
10 |
| 6 | Ph | 10 | PhCF3 | 94 | >100:1 |
51 |
| 7 | Ph | 11 | PhCF3 | 98 | 34:1 |
31 |
| 8 | Ph | 12 | PhCF3 | 88 | >100:1 |
41 |
| 9 | Ph | none | MeTHF | 3 | n.d. | 28 |
| 10 | Ph | 10 | MeTHF | 42 | 17:1 |
52 |
| 11 | Ph | 11 | MeTHF | 26 | 51:1 |
47 |
| 12 | Ph | 12 | MeTHF | 20 | n.d. | 52 |
| 13 | Ph | none | CH2Cl2 | 87 | 26:1 |
6 |
| 14 | Ph | 10 | CH2Cl2 | 99 | 65:1 |
68 |
| 15 | Ph | 11 | CH2Cl2 | 95 | 42:1 |
48 |
| 16 | Ph | 12 | CH2Cl2 | 99 | 84:1 |
64 |
| 17 | 4-MeO-Ph | 10 | CH2Cl2 | 42 | 40:1 |
68 |
| 18 | 4-MeO-Ph | 11 | CH2Cl2 | 53 | 45:1 |
57 |
| 19 | 4-MeO-Ph | 12 | CH2Cl2 | 47 | 36:1 |
74 |
| 20e | 3-NO2-Ph- | 10 | CH2Cl2 | 55 | 9:1 |
54 |
| 21e | 3-NO2-Ph- | 11 | CH2Cl2 | 42 | 7:1 |
41 |
| 22e | 3-NO2-Ph- | 12 | CH2Cl2 | 41 | 7:1 |
81 |
| 23 | 3-NO2-Ph- | none | CHCl3 | 30 | 22:1 |
26 |
| 24f | 3-NO2-Ph- | none | CHCl3 | 9 | 8:1 |
17 |
| 25f | 3-NO2-Ph- | 12 | CHCl3 | 72 | 9:1 |
84 |
| 26f,g | 3-NO2-Ph- | 12 | CHCl3 | 82 | 27:1 |
85 |
The full complexity of identifying this intermediate for the proline catalysed reactions was not apparent when this work started.7a A recent publication by Gschwind et al. that details NMR characterisation of a proline-enamine intermediate highlights some of the difficulties; the intermediate cannot be observed by NMR for ketones, but can be identified at low concentrations for selected aldehydes, using a cryo-probe to assist detection, providing DMSO is used as the solvent.8 However, since no self-assembly occurs in DMSO, we had to detect and identify the enamine in this study in CDCl3.
We investigated the progress of the reaction using ProNap 1 and acetone as electrophile. As expected, we could not detect the enamine intermediate. When acetone was used, two species are present; the new species observed in the 1H-NMR spectrum could be assigned to the corresponding pyrroloimidazolidone 16a7f (Fig. 5, and ESI†). The other species in solution showed the characteristic resonances of ProNap 1, and to confirm that this was 1 and not the corresponding intermediate hemiacetal with hidden methyl resonances, the experiment was repeated using only 0.5 eq of acetone (Fig. 5a). The 1H-NMR spectra showed the same two species observed before but in different relative concentration. Addition of the complementary ureidoimidazole 12 shifted downfield the proton H3 in ProNap 1 compared with the unbound state (Fig. 5c and 5a), which is consistent with the formation of the host–guest pair 1:12. As might be expected given the lack of the NH function, there is no characteristic shift for 16a.
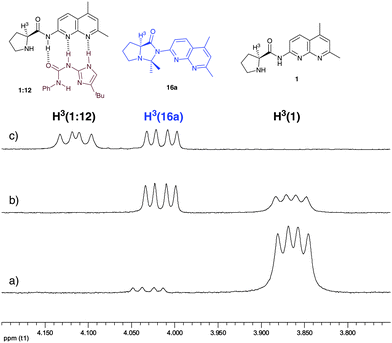 | ||
| Fig. 5 1H-NMR spectra in CDCl3 of the reaction mixture of acetone and ProNap 1 (35 mM) showing the characteristic resonances for Pronap 1 and pyrroloimidazolidone 16a after 2h. a) 0.5 eq. acetone; b) 35 eq. acetone; c) 35 eq. acetone and 1 eq of ureidoimidazole 12. | ||
After addition of acetone, ProNap 1 and pyrroloimidazolidone 16a reach equilibrium, after which the relative concentration of both species remains constant. The relative concentration and the time to reach such equilibrium depends on the concentration of ketone (Fig. 6). More interestingly, the time needed to reach equilibrium can be drastically reduced in the presence of the complementary ureidoimidazole 12, and is reached before NMR analysis can be carried out. In the case of reaction run with 4 eq. acetone, even after 25 h, ProNap and acetone do not yet reach equilibrium or the concentration of 16a observed immediately if ProNap, 12, and acetone are reacted. If solvent (and acetone) is removed from these reactions, the pyrroloimidazolidone starts to revert back to ProNap 1. If pyrroloimidazolidone 16a is in equilibrium with the corresponding enamine, possibly directly, or via the iminium ion, a higher concentration of 16a will “feed” higher concentrations of enamine into solution, and therefore would increase the reaction rate in the enamine catalysed reaction. Since the presence of the complementary co-catalyst will give the higher concentration of intermediate from the start of the reaction, these observations are in accordance with the increase in the reaction rate previously observed in the presence of a complementary co-catalyst to ProNap 1 in the Michael addition reaction between ketones and nitroalkenes. It also explains why the self-assembled catalysts have the unusual ability to deliver good rates without using the large excess of ketone that is more normally employed, since the time taken to reach a reasonable concentration of 16a is prohibitively slow at low ketone concentration. The rate enhancement observed with the co-catalyst is not consistent with the co-catalyst promoting the formation of enamine from 16a, which should be facile process but also would mean the reaction would be zero order in ketone, whereas experiments from this study, our original work and the literature support a positive order in ketone; for example, a comparison of the results in Table 2, entry 23 and 24 in which only the concentration of ketone is altered shows an increase from 9 to 30% conversion in going from 1 equivalent to 4.8 equivalents of ketone. Separate experiments of reaction between β-3-dinitro-styrene and cyclohexanone catalysed by ProNap with co-catalyst 12 in which the concentration of the nitro-styrene was varied show that the reactions are also positive order in nitroalkene (see ESI†). The origin of the rate enhancement with co-catalyst is consistent with increased concentration of 16a feeding active enamine intermediate into the reaction with this concentration of active species and the actual C–C bond forming step being influential on the productivity. The origin of that co-catalytic effect is most readily explained by the co-catalyst promoting the loss of water from the N,O-hemiacetal formed between ketone and ProNap, presumably in a fashion reminiscent of the positive catalytic effect that H-bonding co-catalysts can have on acetalisation.9 It is not clear whether that process is intermolecular using dissociated co-catalyst, or intramolecular, although since a small excess of co-catalyst enhances productivity further, the former may be more likely. In any case, this issue is not central to our main objectives of explaining the unexpected effects of complementary co-catalysts on enantioselectivity or the catalytic results obtained.
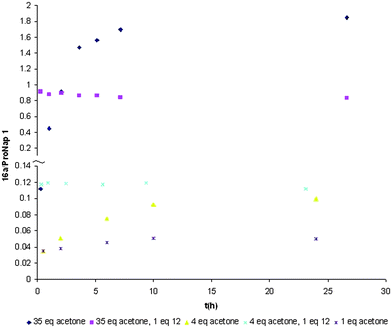 | ||
| Fig. 6 Formation of 16a as a functional of time at different acetone concentrations in presence and absence of cocatalyst 12 (35 mM ProNap in CDCl3). | ||
Next, we attempted to detect the enamine intermediate using an aldehyde. Several aldehydes were considered and partially examined but isovaleraldehyde gave very clear results in the recent publication of Gschwind et al. and therefore enabled us to cross-check our results. ProNap 1 does not catalyse the Michael addition of aldehydes to nitroalkenes very well, even in the presence of co-catalysts, but it does promote the reaction when used stoichiometrically (Table 3, entries 1 and 2). The Michael addition of hexanal and β-nitrostyrene as acceptor using 1 equivalent of ProNap 1 in CDCl3 gives, after 13 min, a small amount of syn-adduct, ca. 4–5% (the characteristic diastereotopic methylene protons next to the NO2 group in the syn-adduct were used for identification purposes). However, no further conversion to Michael products was observed after 0.5 h, 1.5 h, 3.5 h and overnight. These results suggest that a small amount of product is formed at the beginning of the reaction, then the catalyst deactivates. In any case, aldehydes do undergo enamine type reactions using ProNap 1, but just do not turnover well, thus making the study with aldehydes significant to ketone-enamine catalysis.
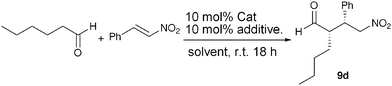
| Entrya | Catalyst | Co-cat. | Conversion [%]b | drc | ee[%]d |
|---|---|---|---|---|---|
| a Reactions carried out at rt in 2 ml of dry CH2Cl2 pre-stirring additive and pre-catalyst for 30 min and using 4.8 eq of hexanal. b Michael products/(Michael products + nitroalkene) × 100 determined by 1H-NMR integration. c Ratio determined by 1H-NMR integration after work-up. d Determined by chiral HPLC. e 70 h. f In MeOH solvent. | |||||
| 1 | (S)-1 | none | Traces | — | — |
| 2 | (S)-1 | 10 | Traces | — | — |
| 3 | (S)-17 | none | Traces e | — | — |
| 4 | (S)-17 | 10 | Traces e | — | — |
| 5 | (S)-18 | none | 6 | n.d. | n.d. |
| 6 | (S)-18 | 10 | 95 | 12:1 |
1 |
| 7f | (S)-18 | none | 99 | 7:1 |
10 |
In order to detect the enamine we prepared a solution of ProNap and isovaleraldehyde (1.5 eq) in CDCl3 (83 mM) and monitored by 1H-NMR spectroscopy (Fig. 7). Initially, there are three different species; one of them in very low concentration, one of the species was a reaction intermediate which disappeared, whilst the concentration of the third species accumulated over time. The assignment of these species as 14, 15 and 16b was made as discussed below.
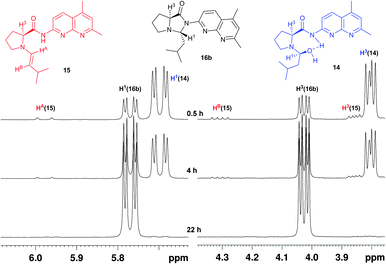 | ||
| Fig. 7 1H-NMR spectra in CDCl3 of the reaction mixture of isobutyraldehyde and ProNap 1 showing the characteristic resonances for the hemiacetal 14 and enamine 15 intermediates and pyrroloimidazolidone 16b. | ||
Identification of enamine 15 (Fig. 7) was quite straightforward due to the characteristic chemical shift and multiplicities of the olefinic protons, 6.02 (HA, d 3JHA,HB = 13.9 Hz), 4.36 (HB, dd 3JHA,HB = 13.9, 3JHB,H4 = 7.0 Hz). This shows an excellent match with data obtained on the elusive enamine intermediate in proline catalysed reactions.8 The size of the coupling constant 3JHA,HB also suggests an E configuration for the enamine. To confirm the structure of enamine 15, a HSQC spectrum was obtained (see Fig. 2b, ESI supporting information†). The doublet of doublet proton resonances at 5.70 and 5.84 ppm assigned to 14 and 16b correlate with sp3 carbons with chemical shifts 75.5 and 79.2 ppm, respectively. On the other hand, the crosspeak of doublet proton resonance at 6.02 ppm in 15 indicates that the corresponding hydrogen is attached to a sp2 carbon with a 13C chemical shift of 132 ppm, which is in accordance with enamine structure, 15.
An expansion of 1H,13C HMBC spectrum of the reaction mixture containing 14, 15 and 16b recorded shortly after mixing reagents in a NMR tube at 273 K in order to obtain higher concentration of intermediate 15 can be found in the Fig. 5b, ESI†. The doublet 1H resonance at 6.02 ppm assigned to 15 shows crosspeaks with Cα, Cδ, CB and C4 (65.2, 51.1, 111.9 and 29.0 ppm, respectively) which confirms connectivity of olefinic moiety with the pyrrolidine ring. A key piece of data for assignment of 16b is the correlation of the doublet of doublet at 5.84 ppm (16b, 1H,13C HMBC) with carbonyl resonance at 176.5 ppm. This is in accordance with imidazolidone ring moiety. An analytically pure sample of N,N acetal 16b, the only isomer observed by 1H-NMR, was obtained by column chromatography on silica gel (EtOAc). The compound was fully characterised (see ESI†) and the relative stereochemistry of pyrroloimidazolidone 16b was determined with the aid of 1D gs-NOESY experiments (Fig. 3, ESI†) and found to be (S, R), as drawn in Fig. 7.
The two main candidates for assigning the third species were N,O hemiacetal 14 or the other diastereomer of 16b (S,S-16b). The iminium ion was felt to be unlikely since H1 appears upfield of the oxazolidinone. The doublet of doublet at 5.70 ppm does not show any crosspeak in the carbonyl region which corresponds with the open structure of intermediate 14 rather than the other possible diastereomer of 16b where the imidazolidone ring is closed. The data obtained is strongly consistent with 14, although not as unequivocal as that which supports 15 and 16b. Further experiments such as an EXSY experiment to confirm that 14 and 15 were in equilibrium were not possible due to the short life-times. It is well established in proline catalysis that the first step before the enamine formation is the addition of the amine to the electrophilic carbon, an aldehyde in this case, forming a N,O hemiacetal that will in turn convert to the corresponding enamine or oxazolidone intermediate,7 in our case a pyrroloimidazolidone. The unusually high field chemical shift for the naphthyridine HD in compound 14 (Fig. 2, compound 1) suggests a strong shielding which tentatively might be rationalised by a strong hydrogen bonding of the amido NH to the hydroxyl group (the same type of shift is calculated for the deprotonated amido-napthyridine that can be envisaged to have similar electron density). This hydrogen bonding may partially assist the assisted elimination of water from hemiacetal 14 and consequent cyclisation to form compound 16b.
Having identified the short-lived enamine intermediate, we investigated if the co-catalyst is hydrogen bonded to the enamine at this stage. Upon addition of the soluble ureidoimidazole 12 to a solution of ProNap 1 in CDCl3 in the presence of isobutyraldehyde (Fig. 8 and Fig. 4, ESI†) a significant NMR shift was observed for the signal assigned to HA on enamine 15, this is consistent with the formation of the host–guest pair 15:12 showing a typical downfield shift for peaks of the species involved in hydrogen bonding compared with the unbound state. This observation strongly suggests that, like ProNap alone, enamine 15 is also hydrogen-bonded to the complementary co-catalyst.
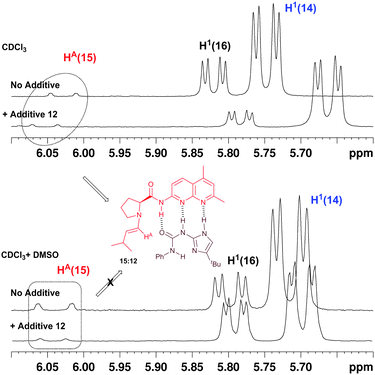 | ||
| Fig. 8 Selected portions of the NMR spectra obtained immediately after addition of iso-valeraldehyde with and without the presence of additive 12 and with and without the presence of DMSO. | ||
In the case of compound 14, the deshielded H1 in the unbound compound moves upfield in the presence of 12. This observation would also fit the tentative proposal made above regarding intramolecular NH-hydroxyl hydrogen bonding that would be disrupted by the co-catalyst, shifting H1 upfield in the NMR spectra. It is worth noting that after 85 min (see Fig. 4 ESI†), compound 16b is almost the only species in solution, suggesting fast formation of enamine 15 and hemiacetal 14 and the corresponding transformation into 16b when the complementary ureidoimidazole is present in solution.
Next, we tried to observe disruption of the hydrogen bonding between the host and the guest pair 15:12 by carrying out the reaction in a strong polar solvent. When the reaction was carried in DMSO or acetonitrile, the only ProNap derived species in solution after 5 min was pyrroloimidazolidone 16b. Since the reaction was too fast to observe any enamine in the reaction mixture, an experiment was carried out in a mixture of CDCl3 and DMSO (0.65 mL + 0.05 mL). The slower reaction rate in chloroform allowed us to observe the presence of enamine 15. In DMSO, the presence of additive 12 did not shift the characteristic enamine HA doublet downfield as it did when a polar solvent is not present (Fig. 8). This observation further supports the formation of the pair enamine-ureidoimidazole 15:12 in solution.
The full conversion of iso-valeraldehyde to pyrroloimidazolidone 16b and the stability of this compound is in strong contrast with the equilibrium between acetone and pyrroloimidazolidone 16a. This difference in stability between imidazolidones derived from aldehydes or ketones strongly supports the possibility that, in contrast with aldehydes, the cyclic pyrroloimidazolidone formed from ketones is less stable and can convert to the corresponding enamine (via iminium ion or directly form the enamine) thus allowing catalysis to take place for ketones but not for aldehydes.
There are reports in the literature where Proline-derived oxazolidinones from ketones have previously been prepared and found to react with different electrophiles (i.e. β-nitrostyrene) to give the corresponding Michael adducts.7b We next investigated if pyrroloimidazolidone 16b derived from isovaleraldehyde and ProNap 1 could catalyse the conjugate addition of acetone to β-3-dinitrostyrene. The reaction was performed in CDCl3 using 4 eq. of acetone, 20 mol% of 16b and was monitored by 1H-NMR (Scheme 1). After 48 h, no new species was detected, showing the stability of 16b under the reaction conditions. Addition of co-catalyst 12, or a small amount of water to promote the hydrolysis of 16b, basically did not change the outcome of the reaction, although traces of a Michael adduct were detected. In contrast, ProNap 1 and ureidoimidazole 12 catalysed the reaction to full conversion giving adduct 9e with a 68% ee.
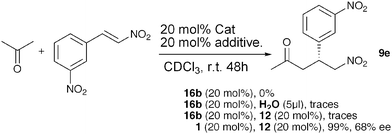 | ||
| Scheme 1 Acetone-Nitro-Michael reaction; comparing pyrroloimidazolidnones with ProNap. | ||
In order to assess if this is a particular behaviour of ProNap 1, another prolinamide lacking the naphthyridine moiety was prepared and tested in the Michael addition of hexanal and β-nitrostyrene (Table 3). Prolinamide 17 did not afford any Michael adduct, with or without the presence of co-catalyst 10 (entries 3 and 4), suggesting that the formation of stable pyrroloimidazolidones from aldehydes is more general for amides derived from proline, and a more general deactivation pathway.
If the formation of pyrroloimidazolidones such as 16b is the only impediment for ProNap 1 catalysing reactions with aldehydes, removing the possibility of intramolecular cyclisation should result in an active catalyst for aldehydes. To study this possibility, β-ProNap, 18 was prepared. The synthesis was performed starting from commercially available (S)-pyrrolidine-3-carboxylic acid10 as described in Scheme 2 (see E.S.I. for full procedures and characterisation).
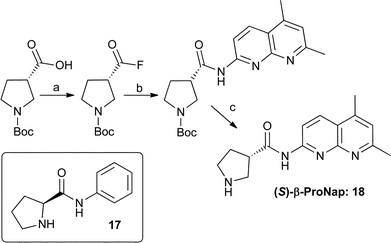 | ||
| Scheme 2 Synthesis of β-ProNap, [(a) Cynanuric Fluoride, CH2Cl2 (quant.), (b) 5,7-dimethyl-1,8-naphthyridin-2-amine, pyridine, DMF (71%) and (c) TFA, CH2Cl2 (93%) and structure of the other Proline-amide investigated for deactivation by aldehydes. | ||
β-ProNap, 18 was next used as catalyst in the nitro-Michael reaction with an aldehyde. β-Pronap should not be capable of forming a stable N,N-acetal, and thus should show some reactivity. Pleasingly, this is the case with a significant enhancement in catalytic activity. β-ProNap, 18 and co-catalyst 10 afforded the Michael adduct 9d in 95% yield in contrast with ProNap 1 (entries 2 and 6). β-ProNap, 18, used alone, also catalysed the full conversion of butyraldehyde into product in MeOH (entry 7). The apparent lack of activity in CH2Cl2 (entry 5), coincides with the very low solubility of compound 18 in CH2Cl2 unless solubising co-catalyst 10 is present, a surprising contrast to the high solubility of ProNap, 1, in this solvent. β-ProNap 18 was also tested in the nitro-Michael reaction with cyclohexanone (Table 1. E.S.I.). The very limited range of complementary co-catalysts invesigated with β-ProNap did not alter the unselective course of these reactions, but to extend the study to bulkier β-proline derived co-catalysts, whether they be receptors for completmentary co-catalysts or not, could be a worthwhile area of study.
Conclusions
In summary, we have shown that ProNap, 1, and complementary ureidoimidazoles form self-assembled catalysts and that the nature of the co-catalyst alters the selectivity of nitro-Michael reactions. We have detected and identified the key enamine intermediate. NMR experiments carried out in CDCl3 also suppport that the co-catalyst is hydrogen bonded at this critical stage. It seems reasonable that the H-bonded enamine would react directly with the nitro-alkenes with the co-catalyst helping to maintain the selectivity of this process. In DMSO, no evidence for H-bonded enamines is observed, which in turn would explain why the positive attributes of the complementary co-catalysts in the Michael addition of ketones to alkenes were cancelled out by the addition of DMSO. The presence of the co-catalyst strongly influences the time needed to reach an equilibrium between ProNap 1 and the ketone derived imidazolidone, shedding light on the faster initial rates observed when the co-catalysts were present in the reaction. Finally, in contrast with ketones, aldehydes form stable pyrroloimidazolidones which are a dead end in the catalytic cycle, therefore explaining why these catalysts give such poor results when aldehydes were used as substrates. This problem, which is more general for Proline-amides is completely alleviated by the use of catalysts derived from (S)-pyrrolidine-3-carboxylic acid. Scheme 3 tentatively suggests the most plausible pathway for these reactions. The subtle interplay of effects which lead to an enhancement of diastereoselectivity and enantioselectivity when the co-catalysts are present represents a very small energy difference in a complex system, and hence a full model would be speculation, and the power of this approach is in identifying avenues to tune catalysts prior to obtaining a complete mechanistic picture. For completeness, a transition state with a bifurcated hydrogen bond promoting approach of the acceptor on the same face as the receptor tag does gives the observed product and does not seem unreasonable.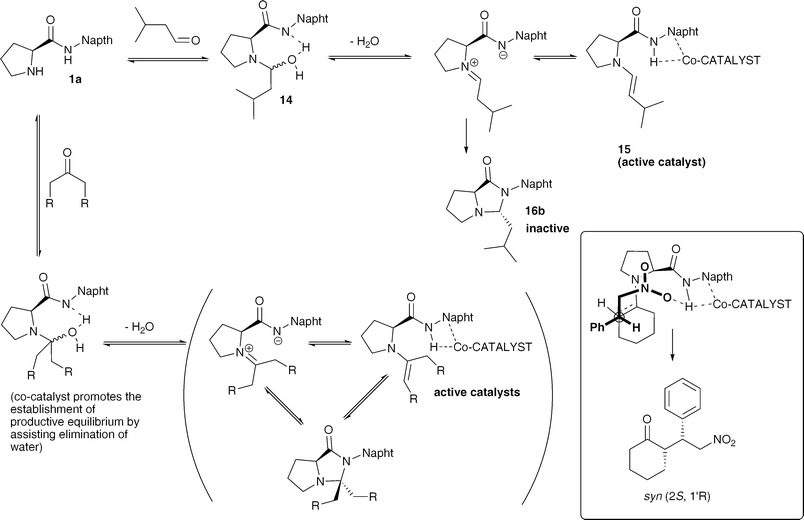 | ||
| Scheme 3 A proposed mechanism and catalytic intermediates for the ProNap catalysed Michael reaction. | ||
The nitro Michael reaction has served as a useful testing ground for examining this approach, but the findings here, are we hope, more generally applicable to conventional enamine catalysis; shedding light on deactivation pathways and catalytic intermediates and the significant equilibria between these. These findings also prove enabling for others working on other types of self-assembled catalysts, representing a rare example of a system that is understood in some detail. We are currently exploring other classes of catalyst including transition metal based systems that can benefit from being programmed to interact with complementary co-catalysts and will report any highlights in due course.
Acknowledgements
We thank the EPSRC for funding for JAF, and for the use of the National Mass Spectroscopy Service. We thanks Prof Doug Philp and Dr Jan Sadownik (University of St Andrews) for some useful discussions and use of curve fitting program for measurement of association constants. Technical support at the University of St Andrews is also gratefully acknowledged.Notes and references
- See for a few arbitrarily chosen examples of organic bifunctional catalysts:
(a) T. Marcelli, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 7496 CrossRef CAS
; (b) H. Miyabe and Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785 CrossRef CAS
- Perspectives on this now burgeoning area:
(a) M. J. Wilkinson, P. W. N. M. Van Leeuwen and J. N. H. Reek, Org. Biomol. Chem., 2005, 3, 2371 RSC
-
(a) M. L. Clarke and J. A. Fuentes, Angew. Chem., Int. Ed., 2007, 46, 930 CrossRef CAS
- For recent reviews and selected publications, see:
(a) T. Mandal and C.-G. Zhao, Angew. Chem., Int. Ed., 2008, 47, 7714 CrossRef CAS
- For recent reviews, see:
(a) S. Sulzer-Mossé and A. Alexakis, Chem. Commun., 2007, 3123 RSC
-
(a) T. R. Kelly, G. J. Bridger and C. Zhao, J. Am. Chem. Soc., 1990, 112, 8024 CrossRef CAS
- For recent publications on the mechanism of proline-catalysed reaction of aldehydes and ketones, see:
(a) B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839 CrossRef CAS
- M. B. Schmid, K. Zeitler and R. M. Gschwind, Angew. Chem., Int. Ed., 2010, 49, 4997 CrossRef CAS
- M. Kolke and P. R. Schreiner, Tetrahedon, 2006, 32, 434
- For pyrrolidine-3-carboxylic acid in enamine-catalysis, see:
(a) H. Zhang, M. Mifsud, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 9630 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, spectroscopic data and selected spectra. CCDC reference number 826224. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00299f |
| This journal is © The Royal Society of Chemistry 2011 |