(C5Me4H)1−-based reduction of dinitrogen by the mixed ligand tris(polyalkylcyclopentadienyl) lutetium and yttrium complexes, (C5Me5)3−x(C5Me4H)xLn†
Thomas J.
Mueller
,
Megan E.
Fieser
,
Joseph W.
Ziller
and
William J.
Evans
*
Department of Chemistry, University of California, Irvine, California +1-92697-2025, U.S.A. E-mail: wevans@uci.edu; Fax: +1-949-824-2210
First published on 3rd August 2011
Abstract
Synthesis of the mixed ligand complexes (C5Me5)(C5Me4H)2Ln (Ln = Lu, Y) for comparison with (C5Me5)2(C5Me4H)Ln to evaluate details of steric effects on reductive reactivity has revealed that (C5Me5)3−x(C5Me4H)xLn complexes can reduce dinitrogen to (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)2−. (C5Me5)(C5Me4H)2Lu reacts with N2 to form [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), (C5Me5)2(C5Me4H)Y reduces N2 to [(C5Me5)2Y]2(μ-η2:η2-N2), and (C5Me4H)3Sc converts N2 to [(C5Me4H)2Sc]2(μ-η2:η2-N2). Exclusive (C5Me4H)1− loss occurs in each case with formation of (C5Me4H)2 as the byproduct. (C5Me5)2, the signature byproduct of sterically induced reduction reactions, is not observed. Since these complexes do not exhibit unusual steric parameters and since the more crowded (C5Me5)2(C5Me4H)Lu and (C5Me5)3Y do not display analogous reactivity, these reactions do not appear to be sterically induced reductions and suggest a new type of ligand-based reduction pathway involving (C5Me4H)1−.
N)2−. (C5Me5)(C5Me4H)2Lu reacts with N2 to form [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), (C5Me5)2(C5Me4H)Y reduces N2 to [(C5Me5)2Y]2(μ-η2:η2-N2), and (C5Me4H)3Sc converts N2 to [(C5Me4H)2Sc]2(μ-η2:η2-N2). Exclusive (C5Me4H)1− loss occurs in each case with formation of (C5Me4H)2 as the byproduct. (C5Me5)2, the signature byproduct of sterically induced reduction reactions, is not observed. Since these complexes do not exhibit unusual steric parameters and since the more crowded (C5Me5)2(C5Me4H)Lu and (C5Me5)3Y do not display analogous reactivity, these reactions do not appear to be sterically induced reductions and suggest a new type of ligand-based reduction pathway involving (C5Me4H)1−.
Introduction
Dinitrogen reduction chemistry using the lanthanide elements was originally developed using complexes of the highly reactive Ln2+ ions, i.e. Sm2+,1Tm2+,2Dy2+,3 and Nd2+,4 as reductants. This area expanded considerably when it was discovered that combinations of trivalent lanthanide complexes with alkali metals could mimic this Ln2+ chemistry and also reduce dinitrogen in reactions designated as LnA3/M where A is an anion that allows these reactions to occur and M is an alkali metal. As shown in Scheme 1, this provided the first reduced dinitrogen complexes of Sc,5 Y,6 and the diamagnetic lanthanides, La7 and Lu.6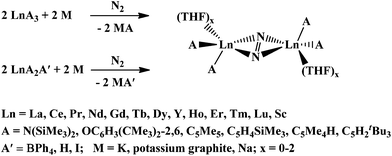 | ||
| Scheme 1 Reduction of dinitrogenviaLnA3/M and LnA2A′/M. | ||
This study was initiated to investigate LnA2A′/M reactions with the mixed ligand species (C5Me5)3−x(C5Me4H)xLn as precursors to determine if mixed ligand [AA′Ln(THF)x]2(μ-η2:η2-N2) products could be obtained. Previously, only [A2Ln(THF)x]2(μ-η2:η2-N2) complexes with identical A ligands were obtainable from LnA2A′/M reactions. The “2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1”8 complexes, (C5Me5)2(C5Me4H)Ln (Ln = Lu, 1; Y, 2) were originally synthesized as shown in eqn (1)9 to probe the subtle details of steric crowding in tris(polyalkylcyclopentadienyl) complexes, (C5Me5)3Ln,10, 11 since (C5Me4H)3Lu,12(C5Me4H)3Y,13 and (C5Me5)3Y14 have been isolated and structurally characterized, but (C5Me5)3Lu has proven elusive. The (C5Me5)2(C5Me4H)Ln compounds were unusual in that they had the bis(pentahapto)-trihapto solid state structure shown in eqn (1).9
:1”8 complexes, (C5Me5)2(C5Me4H)Ln (Ln = Lu, 1; Y, 2) were originally synthesized as shown in eqn (1)9 to probe the subtle details of steric crowding in tris(polyalkylcyclopentadienyl) complexes, (C5Me5)3Ln,10, 11 since (C5Me4H)3Lu,12(C5Me4H)3Y,13 and (C5Me5)3Y14 have been isolated and structurally characterized, but (C5Me5)3Lu has proven elusive. The (C5Me5)2(C5Me4H)Ln compounds were unusual in that they had the bis(pentahapto)-trihapto solid state structure shown in eqn (1).9
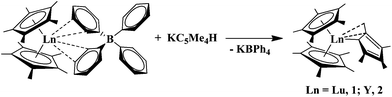 | (1) |
We report here that the LnA2A′/KC8 reactions of these “2:1” (C5Me5)2(C5Me4H)Ln complexes occur with exclusive loss of the (C5Me4H)1− ligand to form [(C5Me5)2Ln]2(μ-η2:η2-N2) and KC5Me4H byproducts. The analogous “1:2” (C5Me5)(C5Me4H)2Ln complexes were synthesized to determine if similar exclusive cleavage of (C5Me4H)1− would occur to form mixed ligand reduced dinitrogen complexes, [(C5Me5)(C5Me4H)Ln]2(μ-η2:η2-N2).
However, in the course of characterizing the (C5Me5)3−x(C5Me4H)xLn complexes it was found that certain metal ligand combinations led to dinitrogen reduction to form trivalent (NN)2− complexes from trivalent tris(polyalkylcyclopentadienyl) precursors in the absence of an external reductant. These reactions were reminiscent of sterically induced reduction reactions of sterically crowded (C5Me5)3Ln compounds, eqn (2), and with
| (C5Me5)3Ln → [(C5Me5)2Ln]+ + ½(C5Me5)2 + 1e1− | (2) |
a growing number of other complexes,15–20 except that the mixed ligand complexes were not sterically crowded and the more crowded (C5Me5)3Ln have never been observed to be strong enough reductants to reduce dinitrogen. These results suggest that a new type of ligand-based dinitrogen reduction reaction exists for (C5Me4H)1− containing complexes. The special nature of the (C5Me4H)1− ligand in dinitrogen chemistry has previously been demonstrated by Chirik et al., but not as a reductant.21, 22
Results
Synthesis of mixed ligand “1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :2” (C5Me5)(C5Me4H)2Ln complexes
:2” (C5Me5)(C5Me4H)2Ln complexes
The “1:2” complexes were synthesized similarly to the eqn (1) synthesis of the “2:1” species (C5Me5)2(C5Me4H)Ln (Ln = Lu, 1, Y, 2). Both (C5Me5)(C5Me4H)2Lu, 3, and (C5Me5)(C5Me4H)2Y, 4, were obtained in >80% yield, eqn (3).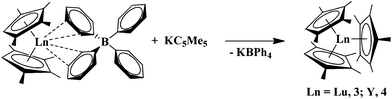 | (3) |
NMR spectroscopy does not indicate if 3 and 4 have the unusual η3-cyclopentadienyl coordination mode observed in 1 and 2, eqn (1). The 1H NMR spectrum of 3 shows one environment for the (C5Me4H)1− ligands and a single resonance for the (C5Me5)1− ligand. This is consistent with all of the cyclopentadienyl ligands bound η5 to the metal in solution. The NMR spectra of (C5Me5)(C5Me4H)2Y, 4, are similar to those of 3 and lack the large Y–C coupling seen in the 13C NMR spectrum of the η3-bound (C5Me5)2(C5Me4H)Y, 2, eqn (1). This suggests that all three cyclopentadienyl ligands in 4 are also bound η5 to the metal. At −50 °C, resonances for two distinct C5Me4H environments are observed for 4, but no evidence of η3-binding is found.
X-ray crystallographic evidence has remained elusive for the “1:2” (C5Me5)(C5Me4H)2Ln complexes 3 and 4. Numerous data sets were collected on these complexes, but the structures could not be solved presumably due to disorder. One single crystal obtained from a solution of 3 gave a structure containing a mixture of (C5Me5)2(C5Me4H)Lu, 1,9 and (C5Me4H)3Lu12 co-crystallized in the same single crystal. As described in a later section, repeated attempts to crystallize 3 and 4 led to unusual dinitrogen reduction chemistry that was recognizable due to the KC8 reactions described in the next section.
Dinitrogen reduction reactivity of the (C5Me5)3−x(C5Me4H)xLn complexes with KC8
The “2:1” complexes, (C5Me5)2(C5Me4H)Lu, 1, and (C5Me5)2(C5Me4H)Y, 2, in the presence of KC8 under a dinitrogen atmosphere produce the reduced dinitrogen complexes [(C5Me5)2Lu]2(μ-η2:η2-N2), 5,23 and [(C5Me5)2Y]2(μ-η2:η2-N2), 6,23 in moderate yields, 49% and 37%, respectively, eqn (4). This is typical LnA2A′/M reductive chemistry according to Scheme 1. These dinitrogen reduction yields are in between those observed when (C5Me4H)3Lu (72%)12 and (C5Me4H)3Y (28%)13 are used as reactants, eqn (5). The byproduct in all of these reactions is exclusively KC5Me4H.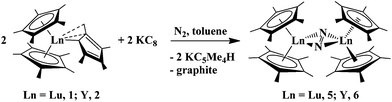 | (4) |
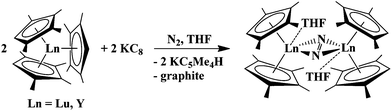 | (5) |
KC5Me4H is also the exclusive byproduct obtained when the “1:2” complex, (C5Me5)(C5Me4H)2Lu, 3, is treated with KC8 under N2. This reaction produces [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), 7, eqn (6), in 23% isolated yield. The ligand redistribution complexes, (C5Me5)2(C5Me4H)Lu, 1, and (C5Me4H)3Lu (major product) were among the other byproducts formed in this reaction.
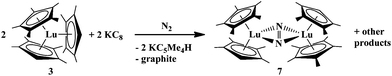 | (6) |
Complex 7, Fig. 1, maintained enough asymmetry to allow the observation of the N–N stretch of the reduced dinitrogen ligand by infrared spectroscopy. In all previous lanthanide reduced dinitrogen complexes of general formula [A2Ln(THF)x]2(μ-η2:η2-N2), no infrared stretch was observed for the (N2)2− moiety. The infrared spectrum of 7 displays an absorption band at 1736 cm−1 that shifts to 1678 cm−1 in the 15N2 labeled analog, 7–1515N (calcd 1677 cm−1 for 15N2). The 1736 cm−1 absorption is attributed to the reduced dinitrogen moiety and is between the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch in free dinitrogen (2359 cm−1)24 and the NN stretch in azobenzene (1482 cm−1).25
N stretch in free dinitrogen (2359 cm−1)24 and the NN stretch in azobenzene (1482 cm−1).25
![ORTEP35 of [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), 7, drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity.](/image/article/2011/SC/c1sc00139f/c1sc00139f-f1.gif) | ||
| Fig. 1 ORTEP35 of [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), 7, drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. | ||
The “1:2” yttrium complex, (C5Me5)(C5Me4H)2Y, 4, in the presence of KC8 under dinitrogen also produced multiple products: at least ten unique (C5Me4H)1− resonances were observed in the 1H NMR spectrum. The major product again was the ligand redistribution product, (C5Me4H)3Y,13 but unlike the analogous lutetium reaction, no mixed ligand reduced dinitrogen complex was isolated or observable by infrared spectroscopy. Upon addition of THF to the reaction mixture, resonances for the previously characterized [(C5Me4H)2Y(THF)]2(μ-η2:η2-N2), 8,13 were also observed in the 1H NMR spectrum, although this is a minor product that also can result from reduction of (C5Me4H)3Y by KC8.13
Reduction of dinitrogen in the absence of an external reductant
The formation of the reduced (N2)2− complexes above was expected based on previously observed LnA2A′/M chemistry.12,23,26 However, formation of (N2)2− complexes by mixed ligand polyalkylcyclopentadienyl complexes was also observed in the absence of KC8. Attempts to crystallize “1:2” (C5Me5)(C5Me4H)2Lu, 3, in C6D6 from an NMR tube stored in a glovebox containing a dinitrogen atmosphere led to a sample exhibiting the resonances for [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), 7, over a period of 3 weeks. Complex 7 can be crystallized from these solutions in 75% yield viaeqn (7), a significantly higher yield than was obtainable viaeqn (6).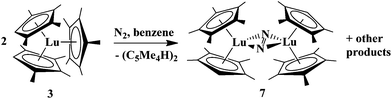 | (7) |
Since the reduction of N2 by 3 alone was such an unexpected result, it was repeated multiple times and the 15N labeled complex 7–1515N was prepared similarly. The NMR, IR, and crystallographic data conclusively show that dinitrogen reduction has occurred. (C5Me4H)2 dimer was observed as a byproduct from this reaction along with resonances for multiple other products. Reactions run on larger scale show formation of red crystalline 7 within a day, but typically two weeks are needed to obtain yields of 50%. Although eqn (7) is slow, it produces 7 in a higher yield and greater purity than the 30 min reactions using KC8 as the reductant, eqn (6).
Other lutetium complexes were examined for similar reactivity, but neither the “2:1” (C5Me5)2(C5Me4H)Lu, 1, nor the “0:3” (C5Me4H)3Lu were observed to reduce N2 under the same conditions. The “1:2” yttrium complex analogous to 3, namely (C5Me5)(C5Me4H)2Y, 4, also did not reduce dinitrogen under analogous conditions.
However, similar ligand-based reactivity was observed from the reactions of “2:1” (C5Me5)2(C5Me4H)Y, 2, and “0:3” (C5Me4H)3Sc, 9, with dinitrogen. Complexes 2 and 9 formed the reduced dinitrogen complexes, [(C5Me5)2Y]2(μ-η2:η2-N2), 6, and [(C5Me4H)2Sc]2(μ-η2:η2-N2), 10, in 51% and 54% crystalline yields, respectively, over a period of 3 weeks, Scheme 2. Again, (C5Me4H)2 was observed as a byproduct.
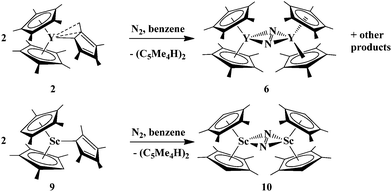 | ||
| Scheme 2 Ligand-based reduction of dinitrogen by tris(polyalkylcyclopentadienyl) complexes, (C5Me5)2(C5Me4H)Y, 2, and (C5Me4H)3Sc, 9. | ||
Structure of [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), 7
Complex 7 has a solid state structure similar to that of [(C5Me4H)2Sc]2(μ-η2:η2-N2), 10,5 in that the cyclopentadienyl ligands have an unusual square planar arrangement. The dihedral angle between the planes defined by the two cyclopentadienyl ring centroids and lutetium for each metallocene component of the molecule is 3.7°, similar to the 2.8° angle in 10. In contrast, the dihedral angles in the unsolvated [(C5Me5)2Y]2(μ-η2:η2-N2), 6,23 and [(C5Me5)2Sm]2(μ-η2:η2-N2)1 complexes are 79.2° and 87.9°, respectively, close to the space efficient 90° angle expected for a tetrahedral arrangement of four cyclopentadienyl ligands around a bimetallic core. In 7, the 2.294 and 2.303 Å Lu–(C5Me4H centroid) distances are indistinguishable from the 2.302 and 2.310 Å Lu–(C5Me5 centroid) distances, Table 1.| 7 | |
|---|---|
| Lu1–Cnt1 (C5Me4H) | 2.303 |
| Lu1–Cnt2 (C5Me5) | 2.310 |
| Lu2–Cnt3 (C5Me4H) | 2.294 |
| Lu2–Cnt4 (C5Me5) | 2.302 |
| Lu1–N1 | 2.291(3) |
| Lu1–N2 | 2.295(3) |
| Lu2–N1 | 2.309(3) |
| Lu2–N2 | 2.308(2) |
| N1–N2 | 1.275(3) |
| Cnt1–Lu1–Cnt2 | 133.4 |
| Cnt3–Lu2–Cnt4 | 134.4 |
The 1.275(3) Å N–N bond distance in 7 is similar to analogs in previously characterized unsolvated complexes of (N2)2−, [A2Ln]2(μ-η2:η2-N2) (A = monoanionic ligand) 1.172(6)–1.259(4) Å,2,5,23,27 as well as the 1.236(8)–1.305(6) Å range for other THF solvated [A2(THF)xLn]2(μ-η2:η2-N2) complexes of this ligand.2–5,7,12,26,27 The 2.291(3)–2.309(3) Å Lu–N distances in 7 are similar to the 2.279(3)–2.292(3) Å Y–N distances in 6 even though Lu3+ is 0.042 Å smaller than Y3+.28
Discussion
The synthesis of the mixed ligand “1:2” (C5Me5)(C5Me4H)2Ln complexes of Lu, 3, and Y, 4, eqn (3), adds to the (C5Me5)3−x(C5Me4H)xLn series (Ln = Lu, Y) in which the total number of methyl substituents can be 12, 13, 14, or 15 on the three rings and the ionic radius of the metal can vary by only 0.042 Å. The subtle effects of steric crowding on structure and reactivity can be readily evaluated with these complexes even though the most crowded member of the series, (C5Me5)3Lu, has not yet been isolated.29
Examination of the (C5Me5)3−x(C5Me4H)xLn series as reactants for LnA3/KC8 and LnA2A′/KC8 reduction of dinitrogen showed a strong preference to remove (C5Me4H)1− instead of (C5Me5)1− in these reactions. The homoleptic (C5Me4H)3Ln (Ln = Lu, Y) complexes had previously been shown to be good reactants for LnA3/KC8 reduction of dinitrogen to form the [(C5Me4H)2Ln(THF)]2(μ-η2:η2-N2) (Ln = Lu8 Y, 813) complexes with formation of byproduct KC5Me4H, eqn (5). Alkali metal reduction of the more crowded “2:1” (C5Me5)2(C5Me4H)Ln (Ln = Lu, 1; Y, 2) complexes also gave KC5Me4H as the byproduct and formed exclusively the bis(pentamethylcyclopentadienyl) reduced dinitrogen complexes, [(C5Me5)2Ln]2(μ-η2:η2-N2) (Ln = Lu, 5; Y, 6), eqn (4).23 No evidence for a mixed ligand reduced dinitrogen complex was observed.
The LnA2A′/KC8/N2 reactions of the “1:2” (C5Me5)(C5Me4H)2Ln complexes were more complicated than those of its less crowded (C5Me4H)3Ln and more crowded (C5Me5)2(C5Me4H)Ln analogs. Multiple products were formed and different results were obtained for lutetiumvs.yttrium.
In the “1:2” (C5Me5)(C5Me4H)2Lu/KC8/N2 reaction, (C5Me4H)1− was again selectively eliminated over (C5Me5)1− and a rare f element example of an (N2)2− complex with two different ancillary ligands on each metal was obtained: [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), 7. This is the first example of a LnA2A′/KC8 reduction in which KA and not KA′ was the byproduct. In 7, this allowed the N–N stretch to be observed by infrared spectroscopy. Another mixed ligand dinitrogen complex predicted to have an IR active NN stretch is [U(η5-C5Me5)(η8-C8H4{SiiPr3-1,4}2)]2(μ-η2:η2-N2), but no absorption assignable to the NN band was observed.30
The “1:2” (C5Me5)(C5Me4H)2Y/KC8/N2 reaction differed from the Lu analog in that no mixed ligand reduced dinitrogen complex was isolated or observed by infrared spectroscopy. The major product of this reaction was the ligand redistribution product, (C5Me4H)3Y. This is understandable since yttrium (1.075 Å 9-coordinate ionic radius)28 is slightly larger than lutetium (1.032 Å)28 and formation of (C5Me4H)3Y is more facile than (C5Me4H)3Lu, which already was the major byproduct in eqn (6). In general, the ease of formation of (C5Me4H)3Ln byproducts may explain the more complicated nature of the “1:2” (C5Me5)(C5Me4H)2Ln/KC8/N2 reactions vs. the “2:1” (C5Me5)2(C5Me4H)Ln/KC8/N2 reactions in which formation of (C5Me5)3Ln would not be favored.
The reduction of dinitrogen by (C5Me5)(C5Me4H)2Lu, 3, without the use of an external reductant, eqn (7), is the most difficult to rationalize. This reduction was unexpected since the more crowded, and presumably more reactive, (C5Me5)3Ln complexes have not been observed to reduce dinitrogen. The observed byproduct from the reduction of N2 by 3, (C5Me4H)2,31 is analogous to the (C5Me5)2 byproduct commonly found in sterically induced reduction (SIR) reactions with the (C5Me5)3Ln complexes,32Scheme 3.
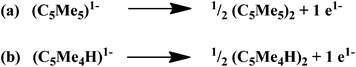 | ||
| Scheme 3 (a) Half reaction of sterically induced reduction (SIR)32, 33 by (C5Me5)3M complexes and (b) the analogous half reaction for (C5Me4H)1−. | ||
However, in SIR reactions, the reactivity is correlated with unusually long M–C(cyclopentadienyl) bond distances.34 Since the structure of 3 is unknown, this could not be evaluated. However, if the reductive reactivity was related to a possibly unusual structure of 3, it did not carry over to the yttrium analog, since (C5Me5)(C5Me4H)2Y, 4, does not show this type of reactivity. Moreover, SIR reactivity increases with steric crowding14,26 and this is not observed in the mixed ligand cyclopentadienyl reductions. Hence, neither of the sterically more crowded molecules, (C5Me5)2(C5Me4H)Lu, 1, and (C5Me5)3Y, reduce dinitrogen. The (C5Me4H)1−reduction is also not specific to either Lu or to “1:2” complexes since “2:1” (C5Me5)2(C5Me4H)Y, 2, and “0:3” (C5Me4H)3Sc, 9, also reduce dinitrogen, Scheme 2. Again (C5Me4H)2 is the observed byproduct. (C5Me5)2(C5Me4H)Y, 2, and (C5Me4H)3Sc, 9, demonstrate that this (C5Me4H)1− reductive reactivity does not require the unusually long bond distances necessary for SIR. Both 2 and 9 have been structurally characterized and have normal structural parameters.9
Finally, since these unusual reductions appear to be effected exclusively with (C5Me4H)1− and not (C5Me5)1− ligands this reactivity appears to be different from SIR. The reductions in eqn (7) and Scheme 1 may involve some as yet unidentified reaction pathways for cyclopentadienyl ligands. This reactivity may involve mono- or tri-hapto coordination modes that are more accessible with (C5Me4H)1− than (C5Me5)1− due to the lower steric congestion at the hydrogen-substituted ring carbon. In any case these results suggest that there are new types of cyclopentadienyl ligand-based reductions beyond SIR and more complexes should be screened with the long reaction times needed to observe this type of unexpected reactivity.
Conclusion
The synthesis of the mixed ligand “1:2” (C5Me5)(C5Me4H)2Ln complexes (Ln = Lu, Y) completes the series of synthetically accessible, closely related, tris(polyalkylcyclopentadienyl) complexes, (C5Me5)3−x(C5Me4H)xLn, that allow steric effects on structure and reactivity to be evaluated by small changes in the number of methyl groups (12–15) and the size of the metal. LnA2A′/KC8/N2 reactions with these complexes gave the first example of a mixed ligand lanthanide complex containing a reduced dinitrogen ligand, [AA′Ln]2(μ-η2:η2-N2), that had an N–N stretch observable by IR spectroscopy. Surprisingly, (C5Me5)(C5Me4H)2Lu, (C5Me5)2(C5Me4H)Y, and (C5Me4H)3Sc were observed to reduce dinitrogen in the absence of an external reductant while their more sterically crowded analogs (C5Me5)2(C5Me4H)Lu and (C5Me5)3Y did not. This suggests that a new type of ligand-based reduction is available from (C5Me4H)1− ligands.
Acknowledgements
We thank the National Science Foundation for support of this research and Ryan A. Zarkesh for assistance with X-ray crystallography.References
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877–6879 CrossRef CAS
.
- W. J. Evans, N. T. Allen and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7927–7928 CrossRef CAS
- W. J. Evans, N. T. Allen and J. W. Ziller, Angew. Chem., Int. Ed., 2002, 41, 359–361 CrossRef CAS
- W. J. Evans, G. Zucchi and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 10–11 CrossRef CAS
- S. Demir, S. E. Lorenz, M. Fang, F. Furche, G. Meyer, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2010, 132, 11151–11158 CrossRef CAS
- W. J. Evans, D. S. Lee and J. W. Ziller, J. Am. Chem. Soc., 2004, 126, 454–455 CrossRef CAS
- W. J. Evans, D. S. Lee, D. B. Rego, J. M. Perotti, S. A. Kozimor, E. K. Moore and J. W. Ziller, J. Am. Chem. Soc., 2004, 126, 14574–14582 CrossRef CAS
- For the purposes of this paper this “#:#” ratio refers to the “number of (C5Me5)1− ligands: number of (C5Me4H)1− ligands.”.
- S. Demir, T. J. Mueller, J. W. Ziller and W. J. Evans, Angew. Chem., Int. Ed., 2011, 50, 515–518 CrossRef CAS
- W. J. Evans, T. J. Mueller and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 2678–2686 CrossRef CAS
- W. J. Evans, T. J. Mueller and J. W. Ziller, Chem. Eur. J., 2010, 16, 964–975 CAS
- W. J. Evans, D. S. Lee, M. A. Johnston and J. W. Ziller, Organometallics, 2005, 24, 6393–6397 CrossRef CAS
- S. E. Lorenz, B. M. Schmiege, D. S. Lee, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 6655–6663 CrossRef CAS
- W. J. Evans, B. L. Davis, T. M. Champagne and J. W. Ziller, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12678–12683 CrossRef CAS
- A. A. Trifonov, E. A. Fedorova, I. A. Borovkov, G. K. Fukin, E. V. Baranov, J. Larionova and N. O. Druzhkov, Organometallics, 2007, 26, 2488–2491 CrossRef CAS
- M. D. Walter, D. Bentz, F. Weber, O. Schmitt, G. Wolmershauser and H. Sitzmann, New J. Chem., 2007, 31, 305–318 RSC
- C. Ruspic, J. R. Moss, M. Schürmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 2121–2126 CrossRef CAS
- P. L. Arnold, E. Hollis, F. J. White, N. Magnani, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 887–890 CrossRef CAS
- R. Jiao, X. Shen, M. Xue, Y. Zhang, Y. Yao and Q. Shen, Chem. Commun., 2010, 46, 4118–4120 RSC
- D. Bojer, A. Venugopal, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2010, 49, 2448–2448 CrossRef
- W. H. Bernskoetter, J. A. Pool, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2005, 127, 7901–7911 CrossRef CAS
- J. A. Pool and P. J. Chirik, Can. J. Chem., 2005, 83, 286–295 CrossRef CAS
- B. M. Schmiege, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 10506–10511 CrossRef CAS
-
K. P. Huber and G. Herzberg, Constants of Diatomic Molecules (data prepared by Gallagher, J.W. & Johnson, R.D. III) in NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P. J.; Mallard, W. G., ed.; National Institute of Standards and Technology: Gaithersburg, MD, 20899. http://webbook.nist.gov (retrieved August 16, 2010) Search PubMed
- A. V. Zemskov, G. N. Rodionova, Y. G. Tuchin and V. V. Karpov, J. Appl. Spectrosc., 1988, 49, 1020–1024 CrossRef
- W. J. Evans, D. S. Lee, C. Lie and J. W. Ziller, Angew. Chem. Int. Ed., 2004, 43, 5517–5519 CrossRef CAS
- F. Jaroschik, A. Momin, F. Nief, X. F. Le Goff, G. Deacon and P. Junk, Angew. Chem. Int. Ed., 2009, 48, 1117–1121 CrossRef CAS
- R. D. Shannon, Acta. Cryst., 1976, A32, 751–767 CrossRef CAS
- W. J. Evans, T. M. Champagne and J. W. Ziller, J. Am. Chem. Soc., 2006, 128, 14270–14271 CrossRef CAS
- F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352–9353 CrossRef CAS
- W. J. Evans, S. A. Kozimor, J. W. Ziller, A. A. Fagin and M. N. Bochkarev, Inorg. Chem., 2005, 44, 3993–4000 CrossRef CAS
- W. J. Evans and B. L. Davis, Chem. Rev., 2002, 102, 2119–2136 CrossRef CAS
- W. J. Evans, J. Organomet. Chem., 2002, 647, 2–11 CrossRef CAS
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Inorg. Chem., 2005, 44, 7960–7969 CrossRef CAS
-
Michael N. Burnett and Carroll K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National Laboratory Report ORNL-6895, 1996 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures, analytical data, X-ray data collection, structure solution and refinement (PDF) and X-ray diffraction details of [(C5Me5)(C5Me4H)Lu]2(μ-η2:η2-N2), 7. CCDC reference numbers 814918–814919. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00139f |
| This journal is © The Royal Society of Chemistry 2011 |