Enantioselective bromination/semipinacol rearrangement for the synthesis of β-bromoketones containing an all-α-carbon quaternary center†
Hui
Li
,
Fu-Min
Zhang
,
Yong-Qiang
Tu
*,
Qing-Wei
Zhang
,
Zhi-Min
Chen
,
Zhi-Hua
Chen
and
Jian
Li
State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, 730000, China. E-mail: tuyq@lzu.edu.cn.; Fax: (+86) 0931-8912582
First published on 7th July 2011
Abstract
A bromination/semipinacol rearrangement reaction catalyzed by cinchona alkaloid derivatives was developed. With 5 mol% (DHQD)2PYDZ, β-bromoketones containing an all-α-carbon quaternary center, which were synthetically useful but challenging to construct, were obtained in up to 97% yield and 93% ee.
The synthesis of all-carbon quaternary centers is a challenge confronting organic chemists and enantioselective construction of this structural motif is of great importance, especially in the context of total synthesis.1 Among the numerous methods that have been developed to reach this target, reactions involving semipinacol rearrangement have proved to be highly efficient.2 However, enantioselective and diastereoselective semipinacol rearrangement is still difficult, particularly in a catalytic fashion.3 From a synthetic point of view, bromonium ion-induced semipinacol rearrangements are of significant importance because the resulting β-bromoketones can undergo further modification.4,5 Recently, our group reported a catalytic asymmetric halogenation/semipinacol rearrangement for the synthesis of α-oxa-quaternary β-haloketones.6b However, catalytic asymmetric construction of all-α-carbon quaternary β-bromoketones is more difficult and has not been reported to date.6
Cinchona alkaloids and their derivatives as organocatalysts have been extensively studied in recent years and have found recent successes in enantioselective halolactonizations.7,8 In connection with our long-standing study of semipinacol rearrangement9 and recent successes in the asymmetric version of this reaction,3e,3f,6 we have attempted to develop an enantioselective bromination/semipinacol rearrangement reaction that is catalyzed by cinchona alkaloid derivatives.
Our hypothesis is depicted in Scheme 1. It was proposed that allylic tertiary alcohol 1 would initially undergo face-selective bromination10 to afford a chiral bromonium ion intermediate that would then simultaneously undergo ring-opening and cause stereospecific migration of R3.
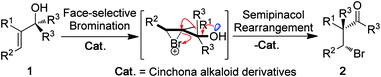 | ||
| Scheme 1 The design of the catalytic asymmetric bromination/semipinacol rearrangement reaction. | ||
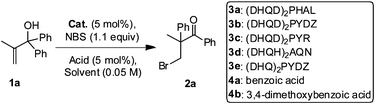
The strategy was first examined by mixing allylic alcohol 1a, hydroquinidine 1,4-phthalazinediyl diether ((DHQD)2PHAL) 3a, N-bromosuccinimide (NBS), and benzoic acid 4a in CH2Cl2 (Table 1, entry 1). Although the desired β-bromoketone 2a was formed in excellent yield, the enantioselectivity of the reaction was very low. After screening some solvents (Table 1, entries 2–4), it was found that toluene and CCl4 inverted the enantioselectivity, while the reaction conducted in CCl4 gave the highest enantioselectivity. Because the reaction proceeded slowly at room temperature in CCl4 (for about 3.5 days), the reaction temperature was increased to 50 °C, which reduced the reaction time to 19 h (Table 1, entry 5). Next, the influence of adding acid, catalyst loading and acid loading on the enantioselectivity of the reaction were investigated (Table 1, entries 6–9). It was found the combination of 5 mol% catalyst and 5 mol% 3,4-dimethoxybenzoic acid 4b gave the best 78% ee (Table 1, entry 8). If no acid was added, the ee decreased to 59% (Table 1, entry 9). In order to further enhance the ee, other catalysts were used (Table 1, entries 10–12). The use of catalyst 3b improved the ee significantly to 87% (Table 1, entry 10).
| Entry | Catalyst | Solvent b | T/°C | Acid | Yield (%)c | ee (%)d |
|---|---|---|---|---|---|---|
| a Reactions were carried out using 5 mol% catalyst, 1.1 equiv. of NBS and 5 mol% acid. b Solvents were used as purchased. c Yield of isolated product. d Determined by chiral HPLC analysis. e With 10 mol% catalyst. f With 1 equiv. of acid. g Dry CCl4 with 5 equiv. of H2O added. h Dry CCl4 with 3 equiv. of CH3OH added. i 1.2 equiv. of NBS was added in six portions (0.2 equiv. every 12 h). | ||||||
| 1 | 3a e | CH2Cl2 | RT | 4a f | 98 | −16 |
| 2 | 3a e | i-PrOH | RT | 4a f | 58 | −21 |
| 3 | 3a e | toluene | RT | 4a f | 55 | 26 |
| 4 | 3a e | CCl4 | RT | 4a f | 51 | 50 |
| 5 | 3a e | CCl4 | 50 | 4a f | 55 | 55 |
| 6 | 3a e | CCl4 | 50 | 4b f | 90 | 63 |
| 7 | 3a | CCl4 | 50 | 4b f | 84 | 65 |
| 8 | 3a | CCl4 | 50 | 4b | 63 | 78 |
| 9 | 3a | CCl4 | 50 | — | 57 | 59 |
| 10 | 3b | CCl4 | 50 | 4b | 69 | 87 |
| 11 | 3c | CCl4 | 50 | 4b | 48 | 30 |
| 12 | 3d | CCl4 | 50 | 4b | 54 | 10 |
| 13 | 3b | CCl4g | 50 | 4b | 77 | 88 |
| 14 | 3b | CCl4h | 50 | 4b | 62 | 90 |
| 15i | 3b | CCl4h | 50 | 4b | 76 | 93 |
| 16i | 3e | CCl4h | 50 | 4b | 59 | −76 |
During the optimization of the reaction conditions, it was discovered that the selectivity decreased when dry CCl4 was used or when 4 Å molecular sieves were added. Therefore, it was speculated that H2O played an interesting role in the reaction. However, deliberate addition of H2O to the reaction (Table 1, entry 13) only caused the ee to increase by 1%. When H2O was replaced with 3 equiv. CH3OH, the ee improved to 90% (Table 1, entry 14). To minimize the background reaction as much as possible, 1.2 equiv. NBS was added to the reaction in six portions (0.2 equiv. every 12 h), which slightly increased the enantioselectivity to 93% ee (Table 1, entry 15). When the quasi-enantiomeric compound 3e was used as the catalyst, the enantiomer of 2a was also isolated in a reduced 59% yield and 76% ee (Table 1, entry 16).
With the optimized conditions for the asymmetric bromination/semipinacol rearrangement in hand (Table 1, entry 15), the scope of the substrate in this reaction was investigated. Different alkyl groups R1 at the C2 position did not influence the efficiency of this reaction. β-Bromoketones 2a–2e were formed in good yield (76–86%) and high enantioselectivity (87–93% ee) (Table 2, entries 1–5). When the migrating aryl group R3 was varied (Table 2, entries 6–11), it was found that both electron-withdrawing and electron-donating substituents were well tolerated. Most allylic alcohols reacted to give the desired products in good yields (62–88%) and selectivities (83–90% ee) (Table 2, entries 6–9), except for 4-fluoro-substituted 1j and bulky 1k, which only gave moderate selectivity (74% ee, 72% ee) (Table 2, entries 10–11). It was found that the substituent R2 at C3 significantly influenced the reaction. When non-terminal olefin 1l was used as a substrate, 2l was generated in only 58% ee (Table 2, entry 12) but with excellent diastereoselectivity. The obtainment of only one single diastereoisomeric β-bromoketone 2l indicated that the ring-opening of bromonium ion and the anti-migration of the phenyl group took place in a perfect concerted process (for additional results with functionalized or beta-branched alkyl groups see the supporting information†).
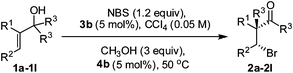
| Entry | Product | Yield (%)c | ee (%)d |
|---|---|---|---|
| a Reactions were carried out on 0.1 mmol scale using dry CCl4 as the solvent. b 0.12 mmol NBS was added in six portions (0.02 mmol every 12 h, reaction time of 72 h). c Yield of isolated product. d Determined by chiral HPLC analysis. | |||
| 1 | 2a: R1 = Me, R2 = H, R3 = Ph | 76 | 93 |
| 2 | 2b: R1 = Et, R2 = H, R3 = Ph | 81 | 91 |
| 3 | 2c: R1 = n-propyl, R2 = H, R3 = Ph | 86 | 90 |
| 4 | 2d: R1 = n-butyl, R2 = H, R3 = Ph | 78 | 91 |
| 5 | 2e: R1 = n-pentyl, R2 = H, R3 = Ph | 83 | 87 |
| 6 | 2f: R1 = Me, R2 = H, R3 = 4-MeC6H4 | 94 | 88 |
| 7 | 2g: R1 = Me, R2 = H, R3 = 3-MeC6H4 | 94 | 90 |
| 8 | 2h: R1 = Me, R2 = H, R3 = 4-ClC6H4 | 62 | 83 |
| 9 | 2i: R1 = Me, R2 = H, R3 = 2-napthyl | 88 | 84 |
| 10 | 2j: R1 = Me, R2 = H, R3 = 4-FC6H4 | 97 | 74 |
| 11 | 2k: R1 = Me, R2 = H, R3 = 3, 5-diMeC6H3 | 89 | 72 |
| 12 | 2l: R1 = Me, R2 = Me, R3 = Ph | 63 | 58 |
To determine the absolute configuration of the products, X-ray crystallographic analysis of 2a was performed. The absolute configuration of 2a was assigned as (S) (Fig. 1)†.
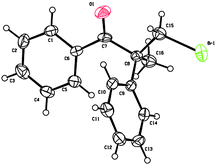 | ||
| Fig. 1 The X-ray crystal structure of 2a. | ||
β-Bromoketones are important building blocks in organic synthesis, so a large-scale reaction is necessary. To evaluate the synthetic utility of this catalytic system, 1 mmol of substrate 1a was used to perform the rearrangement and product 2a was obtained in 74% yield and 91% ee (Scheme 2).
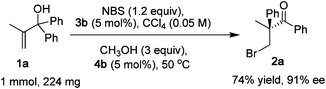 | ||
| Scheme 2 The semipinacol rearrangement of 1a on a 1 mmol scale. | ||
In summary, we developed an efficient and highly enantioselective bromination/semipinacol rearrangement, in which a β-bromoketone containing chiral all-α-carbon quaternary stereogenic center was reported for the first time. Further investigation of the mechanism and synthetic applications of this reaction are in progress.
Acknowledgements
This work was supported by the NSFC (Nos. 20921120404, 20732002, and 20972059), “973” program of 2010CB833200, and the “111” program of MOE.Notes and references
- (a) K. Fuji, Chem. Rev., 1993, 93, 2037–2066 CrossRef CAS; (b) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef; (c) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591–4597 CrossRef CAS; (d) I. Denissova and L. Barriault, Tetrahedron, 2003, 59, 10105–10146 CrossRef CAS; (e) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS; (f) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473–1482 CrossRef CAS; (g) Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed; (h) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS; (i) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS; (j) M. Bella and T. Gasperi, Synthesis, 2009, 1583–1614 CrossRef CAS.
- For reviews see: (a) T. J. Snape, Chem. Soc. Rev., 2007, 36, 1823–1842 RSC; (b) E. Leemans, M. D'Hooghe and N. D. Kimpe, Chem. Rev., 2011, 111, 3268–3333 CrossRef CAS.
- For catalytic asymmetric semipinacol/pinacol rearrangement, see: (a) B. M. Trost and T. Yasukata, J. Am. Chem. Soc., 2001, 123, 7162–7163 CrossRef CAS; (b) B. M. Trost and J. Xie, J. Am. Chem. Soc., 2006, 128, 6044–6045 CrossRef CAS; (c) B. M. Trost and J. Xie, J. Am. Chem. Soc., 2008, 130, 6231–6242 CrossRef CAS; (d) F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS; (e) E. Zhang, C.-A. Fan, Y.-Q. Tu, F.-M. Zhang and Y.-L. Song, J. Am. Chem. Soc., 2009, 131, 14626–14627 CrossRef CAS; (f) Q.-W. Zhang, C.-A. Fan, H.-J. Zhang, Y.-Q. Tu, Y.-M. Zhao, P. Gu and Z.-M. Chen, Angew. Chem., Int. Ed., 2009, 48, 8572–8574 CrossRef CAS; (g) T. Liang, Z. Zhang and J. C. Antilla, Angew. Chem., Int. Ed., 2010, 49, 9734–9736 CrossRef CAS.
- (a) B.-M. Wang, Z.-L. Song, C.-A. Fan, Y.-Q. Tu and W.-M. Chen, Synlett, 2003, 1497–1499 CAS; (b) C.-A. Fan, Y.-Q. Tu, Z.-L. Song, E. Zhang, L. Shi, M. Wang, B.-M. Wang and S.-Y. Zhang, Org. Lett., 2004, 6, 4691–4694 CrossRef CAS; (c) X.-D. Hu, Y.-Q. Tu, E. Zhang, S.-H. Gao, S.-H. Wang, A.-X. Wang, C.-A. Fan and M. Wang, Org. Lett., 2006, 8, 1823–1825 CrossRef CAS; (d) J.-D. Liu, S.-H. Wang, F.-M. Zhang, Y.-Q. Tu and Y.-Q. Zhang, Synlett, 2009, 3040–3042 CAS.
- For bromination/semipinacol rearrangements for the construction of heteroatom-substituted quaternary centers, see: (a) B. Koppenhoefer, K. Lohmiller, F. V. Schurig, in Encyclopedia of Reagents for Organic SynthesisVol. 6, ed. L. A. Paquette, Wiley-VCH, Chichester, 1995, p. 4331 Search PubMed; (b) L. A. Paquette, D. R. Owen, R. T. Bibart, C. K. Seekamp, A. L. Kahane, J. C. Lanter and M. A. Corral, J. Org. Chem., 2001, 66, 2828–2834 CrossRef CAS; (c) P. B. Hurley and G. R. Dake, Synlett, 2003, 2131–2134 CAS; (d) G. R. Dake, M. D. B. Fenster, P. B. Hurley and B. O. Patrick, J. Org. Chem., 2004, 69, 5668–5675 CrossRef CAS; (e) P. B. Hurley and G. R. Dake, J. Org. Chem., 2008, 73, 4131–4138 CrossRef CAS.
- We have previously reported halogenation/semipinacol rearrangements, see: (a) M. Wang, B.-M. Wang, L. Shi, Y.-Q. Tu, C.-A. Fan, S.-H. Wang, X. D. Hu and S.-Y. Zhang, Chem. Commun., 2005, 5580–5582 RSC; (b) Z.-M. Chen, Q.-W. Zhang, Z.-H. Chen, H. Li, Y.-Q. Tu, F.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2011, 133, 8818–8821 CrossRef CAS.
- (a) Y. Chen, S.-K. Tian and L. Deng, J. Am. Chem. Soc., 2000, 122, 9542–9543 CrossRef CAS; (b) S.-K. Tian and L. Deng, J. Am. Chem. Soc., 2001, 123, 6195–6196 CrossRef CAS; (c) S.-K. Tian, Y. Chen, J. Hang, L. Tang, P. McDaid and L. Deng, Acc. Chem. Res., 2004, 37, 621–631 CrossRef CAS; (d) S. J. Connon, Chem. Commun., 2008, 2499–2510 RSC; (e) Cinchona Alkaloids in Synthesis & Catalysis: Ligands, Immobilization and Organocatalysis, ed. C. E. Song, Wiley-VCH, Weinheim, 2009 Search PubMed; (f) L.-Y. Wu, G. Bencivenni, M. Mancinelli, A. Mazzanti, G. bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7196–7199 CrossRef CAS; (g) M. W. Paixão, N. Holub, C. Vila, M. Nielsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 7338–7342 CrossRef.
- (a) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298–3300 CrossRef CAS; (b) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664–3665 CrossRef CAS; (c) G.-F. Chen and S.-M. Ma, Angew. Chem., Int. Ed., 2010, 49, 8306–8308 CrossRef CAS; (d) L. Zhou, C. K. Tan, X. Jing, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474–15476 CrossRef CAS; (e) R. Yousefi, D. C. Whitehead, J. M. Mueller, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 608–611 CrossRef CAS; (f) C. K. Tan, L. Zhou and Y.-Y. Yeung, Org. Lett., 2011, 13, 2738–2741 CrossRef CAS.
- (a) Y.-Q. Tu, L.-D. Sun and P.-Z. Wang, J. Org. Chem., 1999, 64, 629–633 CrossRef CAS; (b) C.-A. Fan, B.-M. Wang, Y.-Q. Tu and Z.-L. Song, Angew. Chem., Int. Ed., 2001, 40, 3877–3880 CrossRef CAS; (c) B.-M. Wang, Z.-L. Song, C.-A. Fan, Y.-Q. Tu and Y. Shi, Org. Lett., 2002, 4, 363–366 CrossRef CAS; (d) X.-D. Hu, C.-A. Fan, F.-M. Zhang and Y.-Q. Tu, Angew. Chem., Int. Ed., 2004, 43, 1702–1705 CrossRef CAS.
- Selected enantioselective bromination reports: (a) A. M. Andrew, A. E. Taggi, H. Wack, J. Esterbrook and T. Lectka, Org. Lett., 2001, 3, 2049–2051 CrossRef; (b) M. Marigo, N. Kumaragurubaran and K. A. Jørgensen, Chem.–Eur. J., 2004, 10, 2133–2137 CrossRef CAS; (c) G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, P. Melchiorre and L. Sambri, Angew. Chem., Int. Ed., 2005, 44, 6219–6222 CrossRef CAS; (d) S. Bertelsen, N. Halland, S. Bachmann, M. Marigo, A. Braunton and K. A. Jørgensen, Chem. Commun., 2005, 4821–4823 RSC; (e) C. Dogo-Isonagie, T. Bekele, S. France, J. Wolfer, A. Weatherwax, A. E. Taggi and T. Lectka, J. Org. Chem., 2006, 71, 8946–8949 CrossRef CAS; (f) P. Goswami, A. Baruah and B. Das, Adv. Synth. Catal., 2009, 351, 1483–1487 CrossRef CAS; (g) T. Kano, F. Shirozu and K. Maruoka, Chem. Commun., 2010, 46, 7590–7592 RSC; (h) K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174–9177 CrossRef CAS; (i) Y. F. Cai, X. H. Liu, Y. H. Hui, J. Jiang, W. T. Wang, W. L. Chen, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2010, 49, 6160–6164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for new compounds. CCDC reference numbers 821778 ((S)-2a). The crystallographic data can be obtained free of charge from The Cambridge Crystallographic Data Centerviahttp://www.ccdc.cam.ac.uk/data_request/cif. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00295c |
| This journal is © The Royal Society of Chemistry 2011 |