Photoinduced reactivity of Au–H intermediates in alcohol oxidation by gold nanoparticles supported on ceria
Andrea
Maldotti
*a,
Alessandra
Molinari
a,
Raquel
Juárez
b and
Hermenegildo
Garcia
b
aDipartimento di Chimica, Università di Ferrara, Via Luigi Borsari 46, 44121, Ferrara, Italy. E-mail: mla@unife.it; Fax: (+39) 0532 240709
bInstituto Universitario de Tecnología Química CSIC-UPV, Universidad Politécnica de Valencia, 46022, Valencia, Spain
First published on 20th June 2011
Abstract
H-Au-np/CeO2 species, that are important intermediates in the alcohol oxidation by gold nanoparticles, show a photoinduced reactivity. UV-visible photoexcitation in deaerated conditions causes a hydride transfer from gold to the spin trap DMPO. The photoassisted breaking of the Au–H bond restores the catalyst in its active form. As a consequence, photochemical excitation has a positive effect on the catalytic oxidation of 2-pentanol under anaerobic conditions, demonstrating that light can be used in place of oxygen. This new photocatalytic activity of H-Au-np/CeO2 species is of general interest in the field of catalysis with gold nanoparticles, since the photoinduced hydride shift from gold surface to organic substrates opens up exciting possibilities to use alcohols for hydrogenation reactions.
Introduction
Oxidation of alcohols for the production of fine chemicals is a demanding transformation.1 In this context, various supported gold catalysts (Au-np) have given very good results.2–5 The first reaction step (Scheme 1) is the formation of a gold-alcoholate species; the subsequent hydride shift to form the carbonylic derivative and an Au–H intermediate is considered the rate determining step.4,5 Finally, the catalytic cycle is closed due to the rapid oxidation of the metal hydride by O2 and the recover of the initial metallic state. This means that the role of oxygen in this mechanism is just that to restore the catalytic activity rather than to oxidize alcohol. The function of the support should be that of providing stability for positive gold species by interfacial metal-support interactions so favouring the formation of Au–H bonds. Addition of base significantly improves the efficiency of the process.6 However, this can also lead to undesired side reactions.7 In the proposed mechanism, the Au–H species is an important intermediate that governs gold activity. Experimental evidence for its formation during the gas phase oxidation of isopropanol and hydrogen spillover on gold nanoparticles supported on ceria (Au-np/CeO2) has been obtained through in situFT-IR studies.8,9 Au–H intermediate formation has also been demonstrated by electron paramagnetic resonance spectroscopy (EPR) and spin trapping technique in toluene and in the absence of base.5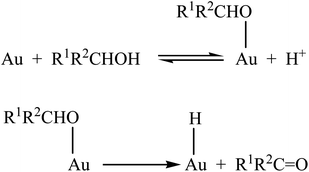 | ||
| Scheme 1 Mechanism of alcohol oxidation by supported Au-np. | ||
It is well known that gold nanoparticles absorb visible light due to the so-called surface plasmon resonance effect (SPR).10–12 It consists in the collective oscillation of conduction electrons that resonate with the electromagnetic field of the incident light. SPR absorption may cause rapid heating of the nanoparticles,13 which can induce oxidation of formaldehyde in air on various oxide supports including CeO2.14Gold nanoparticles also show a considerable UV light absorption causing the transition of 5d electrons to the 6sp band.15,16 It has been reported that supported Au-np can exhibit photocatalytic activity either upon UV or visible light irradiation.17,18 Thus, under UV light irradiation the Au-np on zeolite Y are able to oxidize benzyl alcohol to benzaldehyde with oxygen in the presence of sodium hydroxide in order to increase photocatalytic conversion and selectivity.19
As a part of our ongoing interest in heterogeneous photocatalysis for selective oxidation reactions,20 we are investigating the possibility to employ photoexcited Au-np/CeO2 for catalyzing selective alcohol conversion to carbonylic compounds. Herein, we focus our attention to the possible effect of photoexcitation on the first part of the catalytic mechanism: hydride shift to form H-Au-np/CeO2 species and reactivity of this intermediate. For this reason, we have carried out some EPR spin trapping experiments in the absence of oxygen in order to follow the formation of radical species. In particular, we have irradiated Au-np/CeO2 in the presence of 5,5-dimethylpyrroline N-oxide (DMPO), which is able to trap radicals (R˙) to give more stable nitroxides according to Scheme 2. In some instances, the nature of the trapped radical can be identified by the parameters obtainable from the EPR spectrum.21,22 It is known that spin traps such as DMPO are capable of abstracting intermediates adsorbed over metal surfaces23 or coordinated to a metal center.24EPR-spin trapping technique has been previously employed in order to explore the mechanism of thermal oxidation (70 °C) of alcohols over Au-np/CeO2 catalyst.5
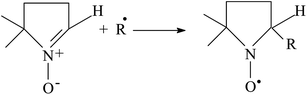 | ||
| Scheme 2 Trapping of a free radical by DMPO. | ||
Results and discussion
The Au-np/CeO2 sample (1.15% w/w) employed in this work corresponds exactly to the same batch that has been previously used to assess the hydrogen spillover and its characterization has been previously reported.9 In short, Au-np/CeO2 is constituted by small gold nanoparticles (average particle size 2.5 nm) supported on nanoparticulate ceria (5 nm mean size). XPS and FT-IR using CO as probe indicate the presence of a population of positive charged gold atoms that are supposed to correspond to coordinatively unsaturated gold atoms located at the corners and edges of the nanoparticles. Fig. 1 reports the diffuse reflectance UV-vis spectrum of the Au-np/CeO2 sample. This spectrum shows a significant ultraviolet absorption and a surface plasmon resonance centered at 560 nm.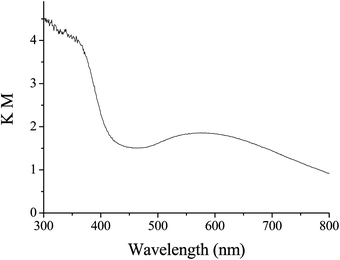 | ||
| Fig. 1 Diffuse reflectance spectrum (plotted as the Kubelka-Munk function) of Au-np/CeO2. | ||
Au-np/CeO2 (5 mgs) was suspended in a mixture of 2-pentanol and toluene containing DMPO. Fig. 2 reports the EPR signal obtained when this sample was left inside the EPR cavity at 70 °C for 20 min under carefully deaerated conditions. The obtained spectrum consists of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet of 1:2:1 triplets with aN = 15.04 G and aH(1) = aH(2) = 19.87 G.
:1:1 triplet of 1:2:1 triplets with aN = 15.04 G and aH(1) = aH(2) = 19.87 G.
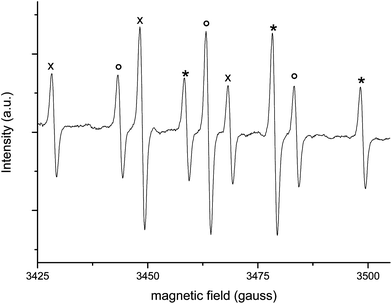 | ||
| Fig. 2 EPR spectrum of the DMPO-H adduct formed in the reaction between Au-np/CeO2 and 2-pentanol in the presence of DMPO (0.05 M) at 70 °C and under anaerobic conditions. | ||
Fig. 3A shows the growth of this signal in time at a fixed-field position (A–B). This result is in agreement with data reported by M. Conte et al.5 In that work, the radical of Fig. 2 was attributed to the adduct DMPO-H, (R = H in Scheme 2). Moreover, isotope labeling experiments and use of a heterogenized nitrone indicated that the spin adduct results from hydrogen abstraction from the H-Au-np/CeO2 species formed as a consequence of hydride shift from alcoholate (Scheme 3 steps a–b).5 Due to the fact that Au–H bond of H-Au-np/CeO2 species is expected to be relatively strong, it is likely that only part of the hydrogen atoms bound onto the gold surface are abstracted by DMPO. Gas chromatographic analysis of the reaction mixture employed for the EPR experiment reveals that 2-pentanol is converted to 2-pentanone, whose yield is reported in Fig. 4. Control experiments indicate that the DMPO-H adduct is not formed in the absence of Au/npCeO2 or alcohol, at room temperature (20 °C), with pristine CeO2.
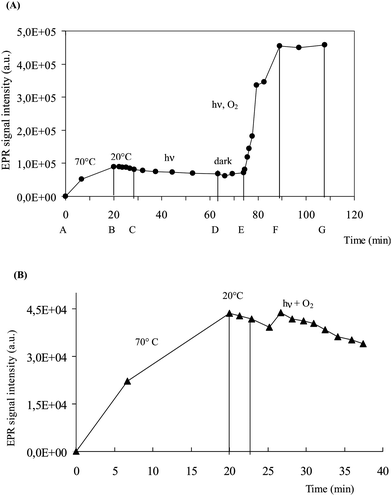 | ||
| Fig. 3 (A) Fixed-field EPR signal intensity of the DMPO-H adduct in time of a suspension of Au-np/CeO2 in a mixture of 2-pentanol and toluene containing DMPO; A–B: 70 °C in the absence of O2, B–C: 20 °C in the absence of O2, C–D: irradiation at 20 °C in the absence of O2, D–E: dark at 20 °C in the absence of O2, E–F: irradiation at 20 °C in the presence of O2, F–G: dark at 20 °C in the presence of O2. (B) fixed-field EPR signal intensity in time during thermal treatment at 70 °C in the absence of O2, in the dark at 20 °C in the absence of O2, irradiation at 20 °C in the presence of O2. | ||
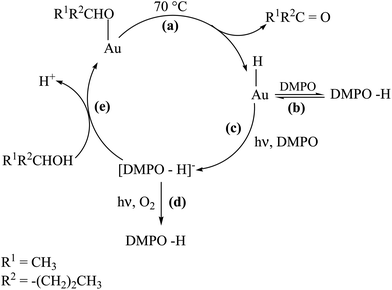 | ||
| Scheme 3 Proposed mechanism for the photoinduced reactivity of Au–H intermediates. | ||
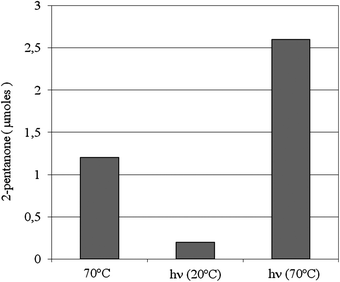 | ||
| Fig. 4 Micromoles of 2-pentanone obtained when Au-np/CeO2 was suspended in a deaerated mixture of 2-pentanol and toluene containing DMPO. Entry 1: 20 min of reaction at 70 °C. Entry 2: 20 min of irradiation (λ > 300 nm) at 20 °C. Entry 3: 20 min of irradiation at 70 °C. | ||
Stability of H-Au-np/CeO2 species is a very crucial point in catalytic oxidation of alcohols, which should cease when these species saturate the surface. For this reason, we investigated the possibility of using UV or visible light to control the reactivity of H-Au-np/CeO2 towards DMPO. Our idea is supported by the findings recently reported by Zhu et al., who demonstrated that illumination can assist the reaction between H-Au-np species on ZrO2 and nitroaromatics.25 After step A–B of Fig. 3A, the subsequent irradiation with light of wavelengths higher than 300 nm at 20 °C do not cause any change of the DMPO-H signal intensity (C–D). On the other hand, an increase of about 6 times is obtained if irradiation is continued upon aerobic conditions (E–F). Control experiments confirm that this enhancement needs the presence of Au-np/CeO2. It is interesting to note that qualitatively similar results are obtained when the sequence of experiments is performed using a filter that cuts off the light of wavelength lower than 450 nm. In these conditions, photochemical excitation occurs only at the plasmon resonance band centered in the visible region (Fig. 1).
Some experiments were carried out in order to verify if photochemical excitation is able to induce the formation of radical species independently of the thermal activation. The results obtained indicate that the DMPO-H adduct is not observed during irradiation at 20 °C of the initial sample in the absence of O2 and without the previous treatment at 70 °C. The EPR spectrum does not appear either when photoexcitation is continued under aerobic conditions. Moreover, the lack of EPR signals due to the trapping of alcohol radicals implies that neither photoassisted electron transfer from alcohol to gold takes place significantly. In line with these findings, the irradiated reaction mixture contained almost negligible amounts of 2-pentanone (Fig. 4).
The growth of the EPR signal observed in the step E–F of Fig. 3A may be explained by assuming that photoexcitation of H-Au-np/CeO2 causes a hydride transfer from gold to the spin trap to give the EPR silent anion DMPO-H− (Scheme 3 step c). This, in turn, should be oxidized to the detected paramagnetic DMPO-H species if irradiation is continued under aerobic conditions (Scheme 3 step d). In fact, it is likely that the photoexcited electrons in gold gain sufficient energy to be captured by oxygen. This should leave positive charges in the metal to oxidize organic molecules such as DMPO-H−.12,25 In line with these statements, it is commonly accepted that paramagnetic spin adducts can also be formed by nucleophilic addition to nitrones followed by oxidation.5 It is seen in Fig. 3B that the EPR signal intensity does not change significantly when photoexcitation under aerobic conditions is carried out immediately after heating at 70 °C, due to the fact that the photoactive intermediates H-Au-np/CeO2 can not be accumulated in the presence of O2.
On the basis of the above considerations, we were intrigued by the possibility that photochemical excitation has a positive effect on the catalytic oxidation of 2-pentanol under anaerobic conditions. In fact, the photoassisted breaking of the Au–H bond should restore the catalyst in its active form also in the absence of O2 (Scheme 3 steps c and e). In accordance with this hypothesis, it is seen (Fig. 4) that, when thermal catalysis and photoirradiation are carried out simultaneously, the micromoles of 2-pentanone are about twice compared to the sum of those obtained in the separate thermal and photochemical experiments.
Working with titania supported gold nanoparticles having gold loading above 20 wt% and large particle size, Kamat and co-workers observed that the energy of the photons absorbed at the gold surface plasmon band can be converted into heat that can even cause the melting of the gold nanoparticles forming larger agglomerates.26 Recently, the temperature dependence of the reaction rate of cumyloxy radical decomposition has been used to estimate that the local temperature at gold nanoparticles in colloidal solutions can be equivalent to a sudden increase of 500 °C for a few microseconds.27 Although some temperature effect upon irradiation of gold nanoparticles cannot be ruled out, it is interesting to note that a comparison of the temperature of a suspension of CeO2 and Au-np/CeO2 (1.15 wt/wt) does not reveal any difference, indicating that, at the macroscopic level, no temperature increase is observed under our irradiation conditions and for our Au-np/CeO2 with low loading and small particle size. Moreover, Fig. 3A (C–D) indicates that a possible temperature increase during irradiation at 20 °C, if it occurs, does not cause, however, appreciable formation of the DMPO-H adduct. Other experimental evidences show that no significant additional effects due to photoinduced temperature enhancement are observed also when irradiation is carried out at 70 °C. In fact, the growth of DMPO-H signal intensity upon simultaneous irradiation and thermal catalysis in the absence of oxygen is similar to that observed upon thermal treatment alone. Therefore, the two-fold increase in 2-pentanone yield obtained when irradiation is coupled to thermal catalysis (Fig. 4) can be finally ascribed to the ability of light of restoring the gold catalyst in its starting state (Scheme 3 steps a, c and e) allowing a new cycle.
Conclusions
The results reported herein confirm that H-Au-np/CeO2 species are important intermediates that govern alcohol oxidation by gold nanoparticles. We have demonstrated that photoexcitation influences the stability of Au–H bonds of these intermediates causing a hydride transfer from gold to the spin trap DMPO with either UV or visible light. We draw attention to the fact that the photoassisted breaking of the Au–H bond restores the catalyst in its active form. As a consequence, photochemical excitation has a positive effect on the catalytic oxidation of 2-pentanol under anaerobic conditions. This new photocatalytic activity of H-Au-np/CeO2 species is of general interest in the field of catalysis with gold nanoparticles. In fact, the feasibility to photoinduce hydride shift from gold surface to organic substrates opens up exciting possibilities to use alcohols for hydrogenation reactions. Future developments may also be possible in the field of hydrogen production.Experimental section
Catalyst preparation: Au-np/CeO2 here employed comes from the same batch of ref. 9 and its preparation is described there.EPR experiments: X band EPR spectra have been recorded using a Bruker 220 D spectrometer. Typical parameters were: frequency 9.75 GHz, power 6.3 mW, sweep width 80 G, center field 3460 G, sweep time 84 s, time constant 81.92 ms, modulation frequency 100 kHz, modulation width 1 G, gain 1.26 × 105. UV-vis spectra have been recorded with a Jasco V-570 spectrophotometer equipped with a diffuse reflectance accessory; the % R values have been transformed via the Kubelka Munk formula.
Samples have been prepared as follows: 0.1 M solution of DMPO in toluene (0.2 mL) was added to neat 2-pentanol (0.2 mL). The dispersing medium was degassed by means of four vacuum line freeze-thaw pump cycles. At the same time, Au-np/CeO2 (5 mg) was left under vacuum for 45 min in order to remove absorbed oxygen; the catalyst was then mixed with the solution under nitrogen atmosphere. The obtained mixture was heated at 70 °C for 20 min. When requested, the sample was irradiated with a Hg medium pressure lamp directly inside the EPR cavity using cut off filters for selecting wavelengths higher than 300 nm or 450 nm. The EPR tube could be opened to the atmosphere, allowing the aeration of the sample. After EPR experiments, the mixture was recovered, the solid was separated and GC analysis of the solution was carried out by using an HP 6890 gas chromatograph equipped with a DB-WAX capillary column (30 m length, 0.32 mm internal diameter, 0.50 μm film thickness). Chromatographic conditions were: 40 °C for 5 min, 10 °C min−1 until 180 °C, carrier flow 1.5 mL min−1, split ratio 10.
Notes and references
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- M. Comotti, C. D. Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS.
- A. Abad, P. Conception, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS.
- A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189–7196 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef.
- A. Abad, P. Conception, A. Corma and H. Garcia, Angew. Chem., 2005, 117, 4134–4137 CrossRef.
- R. Juarez, S. F. Parker, P. Concepcion, A. Corma and H. Garcia, Chem. Sci., 2010, 1, 731–738 RSC.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028–2045 RSC.
- H. Guo, F. Ruan, L. Lu, J. Hu, J. Pan, Z. Yang and B. Ren, J. Phys. Chem. C, 2009, 113, 10459–10464 CrossRef CAS.
- H. Zhu, X. Chen, Z. Zheng, X. Ke, E. Jaatinen, J. Shao, C. Guo, T. Xie and D. Wang, Chem. Commun., 2009, 7524–7526 RSC.
- A. Takami, H. Kurita and S. Koda, J. Phys. Chem. B, 1999, 103, 1226–1232 CrossRef CAS.
- X. Chen, H. Y. Zhu, J. C. Zhao, Z. F. Zheng and X. P. Gao, Angew. Chem., Int. Ed., 2008, 47, 5353–5356 CrossRef CAS.
- C. Voisin, N. Del Fatti, D. Christofilos and F. Valle, J. Phys. Chem. B, 2001, 105, 2264–2280 CrossRef CAS.
- B. Balamurugan and T. Maruyama, Appl. Phys. Lett., 2005, 87, 143105 CrossRef.
- S. Navalon, M. de Miguel, R. Martin, M. Alvaro and H. Garcia, J. Am. Chem. Soc., 2011, 133, 2218–2226 Search PubMed.
- A. Primo, A. Corma and H. Garcia, Phys. Chem. Chem. Phys., 2011, 13, 886–910 RSC.
- J. Zhu, J. L. Figueiredo and J. L. Faria, Catal. Commun., 2008, 9, 2395–2397 CrossRef CAS.
- A. Maldotti and A. Molinari, Topics in Current Chemistry, 2011 Search PubMed , in press.
- E. G. Janzen, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS.
- J. R. Harbour and J. R. Bolton, Biochem. Biophys. Res. Commun., 1975, 64, 803–807 CAS.
- A. Burt, M. Emery, J. Maher and B. Mile, Magn. Reson. Chem., 2001, 39, 85–86 CrossRef CAS.
- C. Bartocci, A. Maldotti, G. Varani, P. Battioni, V. Carassiti and D. Mansuy, Inorg. Chem., 1991, 30, 1255–1259 CrossRef CAS.
- H. Zhu, X. Ke, X. Yang, S. Sarina and H. Liu, Angew. Chem., Int. Ed., 2010, 49, 9657–9661 CrossRef CAS.
- A. Dawson and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 960–966 CrossRef CAS.
- C. Fasciani, C. J. B. Alejo, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, Org. Lett., 2011, 13, 204–207 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |