Organic semiconductor for artificial photosynthesis: water splitting into hydrogen by a bioinspired C3N3S3polymer under visible light irradiation†
Zizhong
Zhang
,
Jinlin
Long
*,
Lifang
Yang
,
Wenkai
Chen
,
Wenxin
Dai
,
Xianzhi
Fu
and
Xuxu
Wang
*
State Key Laboratory Breeding Base of Photocatalysis Fuzhou University, Fuzhou, 350002, P. R. China. E-mail: jllong@fzu.edu.cn; xwang@fzu.edu.cn; Tel: +86 591 83779121
First published on 24th June 2011
Abstract
A novel organic semiconductor photocatalyst mimicking natural light-harvesting antenna complexes in photosynthetic organisms, a disulfide (–S–S–) bridged C3N3S3polymer, was designed and developed to generate hydrogen from water under visible light irradiation. The artificial conjugated polymer shows high H2-producing activity from the half-reaction of water splitting without the aid of a sacrificial electron donor. The H2-producing efficiency and photo-stability of the catalyst could be improved greatly using Ru and single-wall carbon nanotubes as cocatalysts or by adding a sacrificial donor. The results represent a potential and prospective application of the C3N3S3polymer in solar energy conversion and offer significant guidance to develop more stable and efficient photocatalytic systems based on organic semiconductors.
Introduction
Splitting of water into H2 by a photochemical process is one of the biggest chemical challenges in solar energy utilization,1 and thus underpins the intense interest in the creation of new compounds to master “the photosynthetic processes that hitherto have been the guarded secret of the plants”.2 Over the past 30 years, substantial effort has been devoted to developing photofunctional materials that can work under visible light, in order to realize the dream of artificially mimicking photosynthesis to produce hydrogen fuel from water.3 The focus of the search for the visible-light responsive photocatalysts was mainly on inorganic semiconductors including metal oxides (e.g. In1-xNixTaO4, SnNb2O6, PbBi2Nb2O9),4 metal sulfides (e.g. CdS, Zn1-xCuxS, AgInZn7S9),5 metal oxynitrides or oxysulfides (e.g.TiO2-xNx, TaON, LaTiO2N, GaN:ZnO, Sm2Ti2O5S2)6 and some molecular catalysts based on transition metal complexes, such as ruthenium(II)-tris-bipyridine7 and iron hydrogenase enzymes.8 However, these photocatalysts did not always exhibit satisfactory efficiency of H2 production or lifetime. Development of highly efficient visible-light photocatalysts remains one of the focuses of the research on water splitting to hydrogen.Organic conjugated polymers have been widely applied in solar cells and photovoltaic devices,9 because they have commonly a delocalized π electron system and thus can absorb sunlight to create photogenerated charge carriers, and transport these charge carriers. The use of the polymeric materials as a photocatalyst or a co-photocatalyst is attracting increasing attention in the fields of photochemistry and photocatalysis. A few conjugated polymers such as polyaniline, oligo(p-phenylenes), and organic tetracene have been proved to have the potential to function as photocatalysts for the decomposition of organic contaminations or the reduction of CO2 with triethylamine under sunlight illumination.10 The first organic semiconductor reported to allow hydrogen release from water in 1985 was poly(p-phenylenes), but with UV-light as the light source and triethylamine or diethylamine as sacrificial agents.11 Very recently, significant progress in photocatalytic water photosplitting has been made by Wang et al.,12 who reported that a polymeric semiconductor, graphitic carbon nitride (g–C3N4) as a metal-free photocatalyst, has activity for water splitting to H2 under visible-light irradiation with the help of a sacrificial donor. However, to date, no organic semiconductors reported in literature have enabled water to be reduced into hydrogen in the absence of a sacrificial agent under irradiation with visible light. Herein, we drew lessons from photosynthetic biological systems such as algae and foliage to construct a new hydrogen-evolving polymer composed of disulfide (–S–S–) units, which is the pivotal component of light-harvesting complexes in algae and has a strong electron transport capacity,13 and triazine C3N3 ring units. The artificial conjugated polymer can fulfil as a photocatalyst for efficiently generating H2 from pure water under visible-light irradiation without the aid of a sacrificial electron donor.
Results and discussion
This visible-light active polymer photocatalyst was synthesized by a polymerization route in the presence of I2 using 1,3,5-triazine-2,4,6-trithiol as precursor14 (entitled as triazine trithiolpolymer). The combination of XPS and FTIR characterizations (Figure S1 and S2†) with elemental analysis (Table S1†) confirms the disulfide-bridged C3N3S3 structure as depicted in Scheme 1. The XRD pattern presents (Figure S3†) only one broadened Bragg diffraction peak at 22.9°, showing that the resulting solid is an amorphous material. The triazine trithiolpolymer is stable in acid and base solutions. Its ultraviolet-visiblediffuse reflectance spectrum (Fig. 1) clearly indicates an absorption band edge with the semiconducting feature at ca. 515 nm and a moderate tail absorption in the range of 515 to 700 nm. The visible light absorption originates from the formation of the π-electron conjugated chromophores in the polymer, because the precursor unit, sodium triazine trithiol salt monomer, is transparent in the region of 300–800 nm. It is estimated by the equation Eg = 1240/λ on the base of the onset absorption at about 650 nm that the optical band-gap energy of the polymer is ca. 1.91 eV, which is large enough to overcome the thermodynamic barrier of the water-splitting reaction (requiring 1.26 eV theoretically). A broad visible PL band centered at about 550 nm can be seen for the polymer with 400 nm light excitation at room temperature, as shown in Fig. 1 (inset), indicating that the polymer can produce electron-hole pairs upon absorbing photons, which is a crucial step to initialize the photocatalytic reaction.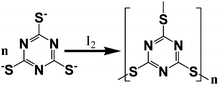
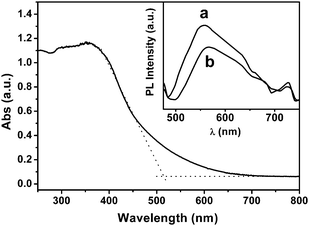 | ||
| Fig. 1 UV-Vis diffuse reflectance spectrum for the as-prepared polymer and wavelength dependence of yield of H2 from the photocatalytic water-splitting over the polymer. The inset shows photoluminescence spectra with 400 nm light excitation at 298 K for polymer (a) and Ru loaded polymer (b). | ||
The density-functional-theory calculations further demonstrate that the triazine trithiol polymer has the potential to function as a photocatalyst for hydrogen production from water, as shown in Fig. 2A presenting the calculated energy levels of the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO). The HOMO orbital mainly consists of antibonding π orbitals of S–S bonds and the LUMO orbital is composed of antibonding σ orbitals of S–S bonds. With increasing polymerization degree (DP) of triazine, the energy level of the HOMO orbital decreases from 1.96 (dimer) to 1.49 eV (hexamer), and the energy level of the LUMO orbital gradually increases from −1.81 (dimer) to −1.34 eV (hexamer). The energy gap between LUMO and HOMO orbitals decreases from 3.77 eV for dimer to 2.83 eV for hexamer. It is expected that the energy gap becomes narrower when the polymerization degree is greater than six. We also performed electrochemical analysis for the as-prepared C3N3S3polymer with a high DP (far greater than six). Fig. 2B shows a typical Mott–Schottky plot of the polymer in the dark, which suggests an n-type feature of the organic semiconductor due to the positive slope of the linear plot.15 Another important parameter derived from the measurement is the flat-band potential of −0.82 V, further confirming the feasibility of photocatalytic water reduction over the polymer. It appears that the LUMO level is more negative than the standard reduction potential of a proton and the HOMO level of +1.09 V is lower than the standard oxidation potential of water, indicating that the reduction reaction of water over the polymer is thermodynamically feasible, while water oxidation over the polymer is infeasible.
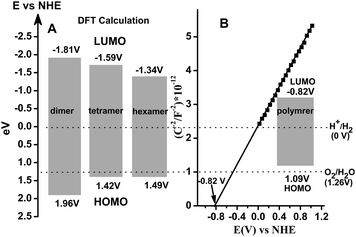 | ||
| Fig. 2 (A) Calculated energy levels of the LUMO and HOMO orbitals for the triazinepolymer with different polymerization degrees. (B) Mott-Schottky plot of the bare C3N3S3polymer. | ||
The above-reported DFT computation and electrochemical analysis are perfectly in line with the catalytic results, as shown in Fig. 3A representing the H2 evolution rate on the triazine trithiol polymer photocatalyst from pure water under visible light illumination (λ ≥ 420 nm). As expected, the as-prepared bare polymer exhibits sustained H2 production from pure water without a sacrificial donor, but no O2 evolution is detected. Water oxidation to O2 evolution by non-metal oxidecatalysts remains a challenge, especially for organic polymercatalysts containing S and N elements due to two aspects: (1) S2− and N3− are more susceptible to oxidation by holes than water;16 (2) O2 evolution has a large over-potential (commonly >0.4 V).17 No O2 evolution leads to the question: what is the half reaction coupled with H2 evolution? XPS analysis (Figure S4†) shows that the disulfide connectors and the triazine C3N3 ring units in the bare polymer could be oxidized by holes to form sulfite (166.5 eV) and nitrite (404.1 eV)18 after a reaction time of 48 h. With increasing reaction time, the triazine trithiol polymer suffers from a slow deactivation for the H2 production from pure water due to the photo-corrosion. In order to avoid the oxidation of anion components in the catalyst, sacrificial agents were required to be injected into the reaction system in general.19 Therefore, we observed the photocatalytic behavior in the presence of some inorganic cation pairs such as Na2S/Na2S2O3, Fe2+/Fe3+, Ce3+/Ce4+; alcohols including methanol and ethanol; and thiocyanate. It was found that both alcohols and Na2S/Na2S2O3 had no effect on the photoactivity of the polymer for H2 evolution. Only Ce3+ and thiocyanate are effective. The use of Ce4+/Ce3+ pair as sacrificial agent can not only increase H2 production from 65 to 80 μmol (Fig. 3A–B),20 but also effectively improves the photo-stability of the polymer as confirmed by the XPS results shown in Figure S5†. No products derived from the photo-corrosion of the polymer, such as sulfite and nitrite, are observed in the presence of the Ce4+/Ce3+ sacrificial agent after the bare polymer has been irradiated with visible-light for 72 h (the discerned sulfate species at 167.8 eV originates from the sacrificial agent). Moreover, we monitor the change of pH value in the reaction system with light irradiation. After adding the Ce3+/Ce4+ coupling pair, the pH value of deionized water decreases from 6.5 to 2.0. But there is no change of pH value during photoreaction of 72 h. Considering that CeO2 was formed as shown by XPS spectrum (Figure S5†) after a reaction time of 72 h in the presence of the Ce4+/Ce3+ sacrificial agent, one can deduce that the improvement of H2 production maybe originates from the consumption of OH− species formed during water photoreduction with Ce4+. Additionally, the formation of CeO2 maybe facilitates the oxidation of Ce3+ by photogenerated holes (ECeO2/Ce3+ of 1.14 V at pH 2.021 is very close to the valence band level (+1.09 V) of the bare polymer), and consequently accelerates the separation of carriers and inhibits the photo-corrosion of catalyst. However, adding Fe2+ causes a slight decrease in H2 production. This is perhaps because iron ions can catalyze the oxidation of the organic polymer.22
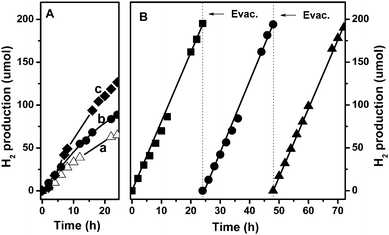 | ||
| Fig. 3 (A) Time courses of visible-light-driven H2 evolution over the polymer from pure water (a), from 2 mmol L−1Ce4+/Ce3+ solution (b), and over the Ru loaded polymer from pure water (c); (B) Time courses of H2 evolution over the Ru loaded polymer in 2 mmol L−1Ce4+/Ce3+ solution under visible light (λ ≥ 420 nm; light intensity: 200 mW cm2; light irradiation area: ca. 28.3 cm−2). | ||
Moreover, the activity of the photofunctional polymer for H2 production can be improved remarkably by loading of co-catalysts such as Ru. As shown in Fig. 3A–C, the photocatalytic activity of the polymer for H2 evolution from water is increased from ca. 65 μmol to ca. 125 μmol as loading 3.0 wt. % Ru. The improvement of H2 production is also achieved using single-wall carbon nanotubes as a cocatalyst or using thiocyanate as a sacrificial electron donor (Figure S5 and S6†). Upon the coexistence of the cocatalyst Ru and the Ce4+/Ce3+ sacrificial donor in the reaction system, H2 is steadily produced over the polymer within 72 h of reaction time, and the rate reaches 8.1 μmol h−1 (Fig. 3B). The overall quantum efficiency (OQE) of the Ru-loaded polymercatalyst is estimated to be 0.023% with irradiation of ≥420 nm (Fig. 3B). We think the enhancing effect of noble metal cocatalysts is derived from two aspects: (1) Noble metal particles deposited on the catalyst surface facilitate the separation and transfer of photogenerated electrons,23 which is confirmed by the facts that the PL signal of the triazine trithiol polymer was suppressed (Fig. 1 inset) and that the measurement of the photocurrent was significantly enhanced (Fig. 5b) in the case of Ru metal. (2) Noble metals can function as efficient H2 evolution promoters for many photocatalysts due to the low over-potential.24
To prove that the reaction is really driven by light absorption, the wavelength dependence of the rate of H2 evolution was examined with band-pass filters of different wavelengths under the condition of fixed light intensity (ca. 10. mW cm−2), as shown in Fig. 4. The three light wavelengths used centered at 360, 400, and 450 nm were distributed, respectively, in the spectral regions of 280–400, 380–425, and 425–470 nm as shown in Figure S8†. The rate of H2 evolution decreases with increasing incident wavelength, matching well with the optical absorption spectrum of the triazine trithiol polymer. According to the amount of evolved H2, the OQE of the bare polymer with irradiation of low light intensity of ca. 10 mW cm−2 are estimated to be 0.20% (280–400 nm), 0.16% (380–425 nm), and 0.08% (425–470 nm), far larger than that (0.023%) of the Ru-loaded polymercatalyst with the help of sacrificial donor and irradiation of high light intensity of ca. 200 mW cm−2 (≥420 nm). This decrease of OQE value with increasing light intensity is consistent with the UV-photocatalysis of TiO2 reported by Ohko and co-workers.25 The inset in Fig. 4 demonstrates the decrease of OQE value with incident wavelength, which is in parallel with the absorption spectrum of the bare polymer. These results confirm that the reaction is indeed a light-driven redox process.
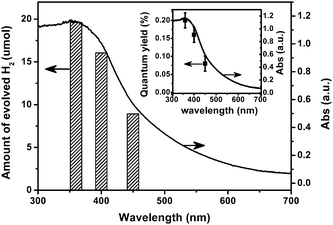 | ||
| Fig. 4 Wavelength dependence of photocatalytic H2 production over the bare C3N3S3polymer. The inset shows the overall quantum yield obtained with irradiation of different incident light wavelengths as a function of absorption spectrum of the bare polymer. The light intensity at various wavelengths was kept at constant (ca. 10 mW cm2) by tuning the current of a Xe lamp. Amount of evolved H2 was obtained without any sacrificial agents after 6 h of irradiation. Light irradiation area is ca. 28.3 cm−2. | ||
Moreover, we use turnover number (TON) to further prove if the reaction proceeds photocatalytically. Considering that the amount of catalyst before and after reaction of 72 h was kept constant (ca. 0.4 g), it was reasonable that surface C3N3S3 units were involved possibly in the reaction, and thus used to calculate TON of the photoreaction. The number of surface C3N3S3 units was estimated to be 4 × 10−4 mol g−1 in the C3N3S3polymer on the base of the specific surface area of the C3N3S3polymer (∼81.6 m2 g−1), which was determined by the Brunauer–Emmett–Teller method at 77 K, and the area of C3N3S3 units (∼0.338 nm2). It can be therefore estimated roughly that TON is approximately 4.0 according to the total H2 evolution of 582 μmol within 72 h as shown in Fig. 3B. The low TON may originate from the low overall photo quantum efficiency (approximately 0.023%) as the result of poor charge separation. This will be further confirmed by photoelectrochemical results later. It can be certain that the amount of hydrogen exceeds that of the photocatalyst significantly, confirming that the reaction is a photocatalytic process. Visible light illumination (λ ≥ 420 nm) of triazine trithiolpolymer layers coated on indium tin oxide (ITO) glass plates reveals a short circuit photocurrent (Fig. 5a), which is still rather low, presumably due to the poor contact between the substrate and the organic powder and the low separation efficiency of the charge carriers. It is interesting to note that the loading of Ru or single-wall carbon nanotubes shows a remarkable improvement in photocurrent, indicating that these cocatalysts promote greatly the efficient separation of photogenerated electron-hole pairs in bulk of the polymer. This is quite in parallel with the photocatalytic activity results reported above. According to these photoelectrochemical results, in combination with the PL results that the charge carriers can be photo-generated with 400 nm light excitation, one can conclude that the artificial conjugated polymer shows a reaction mechanism of water splitting initiated by photogenerated electron-hole pairs, as depicted in the inset to Fig. 5. Upon visible light irradiation, the HOMO electrons jump to the LUMO orbital. The excited electrons can transfer into the catalyst surface to reduce water, while the photogenerated holes are accumulated in the bulk of the catalyst in the absence of a sacrificial agent, causing the self-oxidation of the polymer as confirmed by the XPS results. The presence of the Ce4+/Ce3+ pair not only accelerates H2 production by consuming the concomitant OH− species with Ce4+, but greatly improves the durability of the polymercatalyst by the Ce3+electron donor.
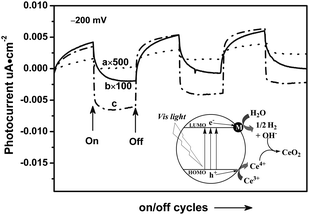 | ||
| Fig. 5 The periodic on/off photocurrent response of the bare polymer (a), the Ru loaded polymer (b) and the single-wall carbon nanotube loaded polymer (c). The inset shows the proposed H2 production mechanism over the C3N3S3polymer under visible light illumination. | ||
The organic semiconductor polymer composed of C3N3S3 units is different structurally from the reported g–C3N4polymer composed of tri-s-triazine units, whereas they display the similar band absorption and the comparable H2 production from water in the coexistence of noble metal cocatalyst and sacrificial agent. It should be noted that compared to the g–C3N4polymer the unmodified C3N3S3polymer has a unique advantage of H2 release in the absence of sacrificial agent, despite the occurrence of photo-corrosion. Further optimization for the H2 evolution system is still needed. Much work involving modification of the catalyst is underway. A preliminary finding reveals that some transitional metal ions such as Cd2+ may be inserted into the disulfide bond of the polymer, causing the enhancement of H2 production (Figure S7†).
Conclusions
In summary, the dream of artificially mimicking photobiological systems to produce hydrogen fuel from water was realized by using a novel organic semiconductor photocatalyst, disulfide (–S–S–) bridged C3N3polymer without the aid of a sacrificial electron donor. The efficiency of H2 production over the polymeric photocatalyst could be improved greatly by adding a cocatalyst or a sacrificial donor. Although the artificial conjugated polymer only shows H2-producing activity from the half-reaction of water splitting, the results represent a potential and prospective application of the organic semiconductor in water photosplitting. This work will enkindle the intense interest in mimicking natural photosynthesis to generate H2 from water.Experimental section
The synthesis of triazine trithiolpolymer: 0.02 mol of tithiocyanuric acid was dissolved in 100 ml of water containing 0.06 mol of NaOH. Then 0.03 mol of iodine in a concentrated aqueous solution of KI was slowly added at 0 °C and the mixture was stirred overnight. A yellowish precipitate was washed with deionized water several times to remove residual ions, and finally dried under vacuum at 60 °C.Characterization of catalysts: The UV/Vis spectra were recorded on a Varian Cary 500 Scan UV/Vis system. The XPS was carried out on an ESCALAB 250 XPS System with a monochromatized Al Kα X-ray source (15 kV, 200 W, 500 μm, pass energy = 20 eV). PL spectra were surveyed by an Edinburgh FL/FS900 spectrophotometer. FTIR spectra were recorded using a Nicolet Magna 670 FTIR spectrometer with a DTGS detector. The N2 absorption-desorption isotherms were determined at 77 K with a Micromeritics ASAP 2010 instrument.
Preparation of the triazine trithiol polymerelectrode: The working electrode was prepared on indium-tin-oxide (ITO) glass plates. The ITO glass pieces with a size of 5 × 1.0 cm were sonicated in acetone and ethanol, and then rinsed with deionized water and dried in an air stream. 25 mg of the powder was ground in a porcelain mortar with 0.5 ml of water to produce slurry. The obtained slurry was used for spreading onto the ITO glass substrate, whose side part was previously protected using Scotch tape. The paste was dried at room temperature as a working electrode before measurement. A copper wire was connected to the side part of the ITO glass using a conductive tape. Uncoated parts of the electrode were isolated with epoxy, and the exposed area of the electrode under illumination was 0.25 cm2.
Photoelectrochemical measurements: visible light photocurrent was measured using the conventional three electrode photoelectrochemical cell. The working triazine trithiolpolymerelectrode was immersed in a sodium sulfate electrolyte solution (0.2 M) (pH ≈ 6.8) with a platinum foil counter electrode and an Ag/AgCl reference electrode. The working electrode was irradiated from the back side (substrate/semiconductor interface) in order to minimize the influence of thickness of the semiconductor layer. The electrode was excited by a 300 W xenon lamp with an UV (≥420 nm) cut-off filter and an IR cut-off filter and was chopped manually. The light/dark short circuit photocurrent response under −0.2 V bias was recorded with a BAS Epsilon Electrochemical Workstation. Mott-Schottky experiments were also carried out with a platinum foil counter electrode and an Ag/AgCl reference electrode in a sodium sulfate electrolyte solution (0.2 M) (pH ≈ 6.8), the potential ranged from −0.2 V to 0.8 V, and the perturbation signal were 10 mV with the frequency at 1KHz.
Photocatalytic activity testing: Photocatalytic reactions were conducted in a gas-closed circulation system with a side window Pyrex cell. H2 production was studied on a dispersion of 0.4 g of catalyst in 400 ml of deionized water. Ru cocatalysts were loaded by an in situphotodeposition method with aqueous solution of RuCl3. The reaction system was evacuated several times by a mechanical pump to remove air. The reaction system was circulated with a glass piston pump. The photocatalyst was dispersed in the water by stirring with a magnetic stirrer and irradiated under a 300 W Xe lamp with a 420 nm cut-off filter or a band-pass filter. The amount of H2 evolved in the circulated system was determined by an online gas chromatograph (HP7890, TCD, Ar carrier).
Acknowledgements
This work was financially supported by the NSFC (Grants Nos. 21003021, 21033003, and 20873022), National Basic Research Program of China (973 Program, No 2007CB613306), the National High Tech R&D Program of China (863 Program, 2008AA06Z326), the Natural Science Foundation of Fujian Province of P.R. China (2010J05024), and the Science and Technology Project of Education Office of Fujian Province of P.R. China (JK2010002).References
- (a) K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS; (b) R. M. Navarro, M. A. Pena and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952 CrossRef CAS; (c) R. M. Navarro, M. C. Sánchez-Sánchez, M. C. Alvarez-Galvan, F. Valle and J. L. G. Fierro, Energy Environ. Sci., 2009, 2, 35 RSC; (d) J. A. Turner, Science, 2004, 305, 972 CrossRef CAS; (e) X. L. Fu, J. L. Long, X. X. Wang, D. Y. C. Leung, Z. X. Ding, L. Wu, Z. Z. Zhang, Z. H. Li and X. Z. Fu, Int. J. Hydrogen Energy, 2008, 33, 6484 CrossRef CAS.
- (a) A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022 CrossRef CAS; (b) G. Ciamician, Science, 1912, 36, 642 Search PubMed; (c) A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS; (d) M. Ni, M. K. H. Leung, D. Y. C. Leung and S. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401 CrossRef CAS; (e) M. Matsuoka, M. Kitano, M. Takeuchi, K. Tsujimaru, M. Anpo and J. M. Thomas, Catal. Today, 2007, 122, 51 CrossRef CAS.
- (a) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC; (b) K. Domen and K. Maeda, J. Phys. Chem. C, 2007, 111, 7851 CrossRef; (c) F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- (a) R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS; (b) Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625 CrossRef CAS; (c) Y. Hosogi, K. Tanabe, H. Kato, H. Kobayashi and A. Kudo, Chem. Lett., 2004, 33, 28 CrossRef CAS; (d) H. G. Kim, D. W. Hwang and J. S. Lee, J. Am. Chem. Soc., 2004, 126, 8912 CrossRef CAS.
- (a) N. Buehler, K. Meier and J. F. Reber, J. Phys. Chem., 1984, 88, 3261 CrossRef CAS; (b) A. Kudo and M. Sekizawa, Catal. Lett., 1999, 58, 241 CrossRef CAS; (c) A. Kudo, I. Tsuji and H. Kato, Chem. Commun., 2002, 1958 RSC; (d) I. Tsuji, H. Kato, H. Kobayshi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406 CrossRef CAS.
- (a) G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698 RSC; (b) A. Kasahara, K. Nukumizu, G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. A, 2002, 106, 6750 CrossRef CAS; (c) K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286 CrossRef CAS; (d) A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547 CrossRef CAS.
- J. G. Vos and J. M. Kelly, Dalton Trans., 2006, 4869 RSC.
- L. Florin, A. Tsokoglou and T. Happe, J. Biol. Chem., 2001, 276, 6125 CrossRef CAS.
- (a) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef; (b) J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1708 Search PubMed; (c) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS; (d) C. J. Brabec, Sol. Energy Mater. Sol. Cells, 2004, 83, 273 CrossRef CAS.
- (a) D. Mahanta, G. Madras, S. Radhakrishnan and S. Patil, J. Phys. Chem. B, 2008, 112, 10153 CrossRef CAS; (b) H. Y. Kim, T. G. Bjorklund, S.-H. Lim and C. J. Bardeen, Langmuir, 2003, 19, 3941 CrossRef CAS; (c) B. Muktha, G. Madras, T. N. G. Row, U. Scherf and S. Patil, J. Phys. Chem. B, 2007, 111, 7994 CrossRef CAS; (d) S. Matsuoka, T. Kohzuki, C. Pac, A. Ishida, S. Takamuku, M. Kusaba, N. Nakashima and S. Yanagida, J. Phys. Chem., 1992, 96, 4437 CrossRef CAS; (e) H. Y. Kim, T. G. Bjorklund, S.-H. Lim and C. J. Bardeen, Langmuir, 2003, 19, 3941 CrossRef CAS.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474 RSC.
- (a) X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS; (b) K. Maeda, X. C. Wang, Y. Nishihara, D. L. Lu, M. Antonietti and K. J. Domen, J. Phys. Chem. C, 2009, 113, 4940 CrossRef CAS; (c) X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS.
- F. Katzen and J. Beckwith, Cell, 2000, 103, 769 CrossRef CAS.
- F. J. Cataldo, J. Inorg. Organomet. Polym., 1997, 7, 35 CrossRef CAS.
- D. S. Kong, Langmuir, 2008, 24, 5324 CrossRef CAS.
- (a) A. B. Ellis, S. W. Kaiser, J. M. Bolts and M. S. Wrighton, J. Am. Chem. Soc., 1977, 99, 2839 CrossRef CAS; (b) R. J. Williams, Chem. Phys., 1960, 32, 1505 CAS; (c) K. Maeda, N. Saito, D. Lu, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 4749 CrossRef CAS; (d) R. Nakamura and Y. Nakato, J. Am. Chem. Soc., 2004, 126, 1290 CrossRef CAS.
- H. P. Maruska and A. K. Ghosh, Sol. Energy, 1978, 20, 443 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol, K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Physical Electronic, Inc., USA, 1992 Search PubMed.
- (a) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC; (b) M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401 CrossRef CAS.
- E. A. Kozlova, T. P. Korobkina and A. V. Vorontsov, Int. J. Hydrogen Energy, 2009, 34, 138 CrossRef CAS.
- R. Chi, Z. Li, C. Peng, H. Gao and Z. Xu, Metall. Mater. Trans. B, 2006, 37B, 155 CrossRef CAS.
- M. Prudent and H. H. Girault, Metallomics, 2009, 1, 157 RSC.
- (a) M. Hara, J. Nunoshige, T. Takata, J. N. Kondo and K. Domen, Chem. Commun., 2003, 3000 RSC; (b) J. Sato, H. Kobayashi, K. Ikarashi, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2004, 108, 4369 CrossRef CAS.
- (a) S. K. Lee and A. Mills, Platinum Met. Rev, 2003, 47, 61 CAS; (b) W. X. Dai, X. X. Wang, P. Liu, Y. M. Xu, G. S. Li and X. Z. Fu, J. Phys. Chem. B, 2006, 110, 13470 CrossRef CAS.
- Y. Ohko, K. Hashimoto and A. Fujishima, J. Phys. Chem. A, 1997, 101, 8057 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XPS and FTIR characterizations with elemental analysis of the prepared disulfide-bridged C3N3S3polymer (Figure S1, S2 and Table S1). XRD and SEM patterns of the as-prepared C3N3S3polymer (Figure S3). The XPS characterization for the polymer after photocatalytic reaction in pure water or in Ce4+/Ce3+ solution (Figure S4). Time courses of visible-light-driven H2 evolution over single-wall carbon nanotube loaded polymer from pure water (Figure S5). Time courses of visible-light-driven H2 evolution over single-wall carbon nanotube loaded polymer from pure water over Ru loaded polymer from water containing thiocyanate (Figure S6), over Cd3(C3N3S3)2catalyst from pure water (Figure S7). Spectral distributions of the band-pass filters (Figure S8). The detailed description of Density Functional Theory Calculations (DFT) for the LUMO and HOMO orbitals of the polymer (Table S2). See DOI: 10.1039/c1sc00257k |
| This journal is © The Royal Society of Chemistry 2011 |