Distinct conformational preferences of prolinol and prolinol ether enamines in solution revealed by NMR†
Markus B.
Schmid
,
Kirsten
Zeitler
and
Ruth M.
Gschwind
*
Institut für Organische Chemie, Universität Regensburg, D-93040, Regensburg, Germany. E-mail: ruth.gschwind@chemie.uni-regensburg.de; Fax: (+49) 941 943-4617; Tel: (+49) 941 943-4625
First published on 6th July 2011
Abstract
Enamines, which are key intermediates in organocatalysis derived from aldehydes and prolinol or Jørgensen–Hayashi-type prolinol ether catalysts, were investigated conformationally in different solvents by means of NMR spectroscopy, in order to provide an experimental basis for a better understanding of the origin of stereoselection. For all of the enamines studied, surprisingly strong conformational preferences were observed. The enamines of the diarylprolinol (ether) catalysts were found to exclusively exist in the s-trans conformation due to the bulkiness of the pyrrolidine α-substituent. For prolinol enamines, however, a partial population of the s-cis conformation in solution was also evidenced for the first time. In addition, for all of the enamines studied, the pyrrolidine ring was found to adopt the down conformation. Concerning the exocyclic C–C bond, the sc-exo conformation, stabilized by CH/π interactions, is exclusively observed in the case of diarylprolinol ether enamines. In contrast, diarylprolinol enamines adopt the sc-endo conformation, allowing for an OH⋯N hydrogen bond and a CH/π interaction. A rapid screening approach for the different conformational enamine features is presented and this was applied to show their generality for various catalysts, aldehydes and solvents. Thus, by unexpectedly revealing the pronounced conformational preferences of prolinol and prolinol ether enamines in solution, our study provides the first experimental basis for discussing the previously controversial issues of s-cis/s-trans and sc-endo/sc-exo conformations. Moreover, our findings are in striking agreement with the experimental results from synthetic organic chemistry. They are therefore expected to also have a significant impact on future theoretical calculations and synthetic optimization of asymmetric prolinol (ether) enamine catalysis.
Introduction
In-depth studies on intermediate species are highly important for a better understanding of the mechanistic principles that underlie organic reactions. In particular, in the important and ever growing field of stereoselective catalysis, conformational analyses of active intermediates may guide researchers towards the origin of stereocontrol in asymmetric reactions and are, therefore, highly valuable for the directed optimization of already existing catalysts and the design of novel high-performance catalysts. Modern asymmetric organocatalysis,1–5 with its manifold different concepts and activation modes,6,7 such as non-covalent catalysis via phase transfer,8 or hydrogen bonding,9–11 or Brønsted acids12,13 as well as covalent catalysis via Lewis bases,14 has substantially contributed to the field of stereoselective catalysis during the last few years. Typically by making use of compounds originating from the chiral pool, catalysis by secondary amines15–17 through enamine,18,19 iminium,20,21 or SOMO22–24activation has emerged as one of the most successful and widely applicable principles. In particular, proline25–27 and Jørgensen–Hayashi-type prolinol ethers28–33 have been proven to give remarkable performances in asymmetric iminium and enamine organocatalysis. Also, prolinol organocatalysts34 have found a use based on enamine intermediates,35–38 although they are mainly employed in iminium catalysis.However, regarding the vast number of synthetic applications, conformational studies on enamine intermediates, especially on the origin of their stereoselection, are rather scarce and experimental investigations in solution are completely nonexistent so far. This can be partially ascribed to the currently limited number of reports on relevant enamines in solution; no more than two prolinol silyl ether-type enamines have been isolated and characterized,39,40 while only one dienamine intermediate41 and one product enamine42 have been observed in situ. Therefore, the conformations of such enamine intermediates in solution are largely unknown and conformational information has, so far, been limited to theoretical calculations39b,41,43–45 and crystal structure analyses39 . However, these approaches may be affected by vacuum calculation artifacts or crystal packing effects. Accordingly, conflicting results concerning the conformational preferences of both the exocyclic N–C bond39,41,43–46 and the exocyclic C–C bond39b,41,43–45 of diarylprolinol ether enamines have been reported from these studies. Therefore, experimental results in solution are highly desirable to clarify these issues. Only recently have we expanded the available pool of enamines in solution by the first enamine intermediates derived from proline47 and prolinols46 and by various aldehyde-derived prolinol ether enamines.46 Thus, the experimental basis is available for more detailed conformational studies on enamine intermediates in solution. This should help to clarify the origin of stereoselection and, hence, to tailor optimized organocatalysts.
In this article, we present the first detailed in situ investigations on the conformations of aldehyde-derived prolinol and prolinol ether enamines in different solvents by means of NMR spectroscopy. 1H,1H-NOESY spectra reveal the preference of the enamine s-trans arrangement due to the steric influence of the pyrrolidine α-substituent. In addition, the pyrrolidine ring was shown by scalar coupling constants to predominantly adopt the down conformation, which allows for intramolecular CH/π interactions between pyrrolidine protons and the aryl groups of the “obese” α-substituent. In the case of diarylprolinol ether enamines, the sc-exo conformation for the exocyclic Cα–Cε bond was exclusively observed, which is stabilized by two CH/π interactions. In contrast, for the diarylprolinol-derived enamines, only the sc-endo conformation was found, which allows for both an OH⋯N hydrogen bond and one CH/π interaction. In addition, we present a rapid and facile 1D 1H NMR-based screening approach for this conformational feature that plays a key role in the shielding of one face of the enamine and, hence, in the stereocontrol effectuated by the organocatalyst.
Results and discussion
Model enamines
On the basis of our recent studies on proline enamines47 and on the formation and stability of prolinol (ether) enamines46 in solution, various typical secondary amine organocatalysts (Scheme 1: 1–7)28–30 were selected for our conformational enamine study. Two different aliphatic aldehydes with alkyl chains of different sizes, 3-methyl-butyraldehyde a and propionaldehyde b (Scheme 1), were chosen with regard to the suppression of the self-aldolization (a)47 and to a substantial relevance for synthetic applications (b). To allow for the comparison of prolinol and prolinol ether enamines, we predominantly used DMSO in our conformational investigations, since it is the only solvent in which prolinol enamines have been detected so far.46 For prolinol ether enamines, further solvents (methanol, acetonitrile, chloroform, dichloromethane, and toluene) were then used to explore the generality of the conformational preferences. | ||
| Scheme 1 A) Aldehydes and organocatalysts studied. B) Atom nomenclature used for the respective enamines. | ||
All of the experiments were conducted in NMR tubes by mixing equimolar amounts of the aldehyde and catalyst in deuterated solvents in order to obtain concentrations of 50 mmol L−1 each and NMR spectra were recorded at 300 K (see the supplementary information for details†). Overall, 14 different enamines were formed from the aldehydes a–b and the organocatalysts 1–7 (designated as “catalyst-number.aldehyde-character”, i.e.1a–7b in Scheme 2) were obtained in situ and investigated in different solvents. The detection and characterization as mainly E-configured enamines has been reported recently.46 (See also Schemes S1 and S2 in the supplementary information for the NMR assignments†.)
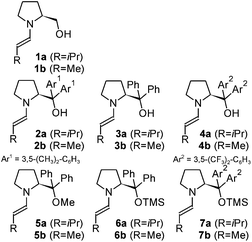 | ||
| Scheme 2 The investigated E-enamines derived from aldehydes a and b and catalysts 1–7, displayed in the favorable s-trans conformation. | ||
Enamine conformations
Besides the already reported aspects concerning the stability, formation and degradation of enamine intermediates,46 knowledge on how the stereoselectivity is controlled in the bond-forming step is of utmost importance for the understanding of asymmetric organocatalyzed reactions. On the basis of previous theoretical calculations39b,41,43–45 and crystal structure analyses,39 and in agreement with previous experimental results, it is generally assumed that the methanol(ether)-substituent of the pyrrolidine ring secures both the s-trans arrangement of the enamine and the effective shielding of one face of the enamine π system, thereby directing incoming electrophiles to the opposite side.39b In the following paper, we present the results of our conformational investigations of the prolinol (ether) E-enamines 1a–7b in solution by NMR spectroscopy, mainly by 1H,1H-NOESY spectra. Addressing the conformational preferences of the pyrrolidine ring and of the exocyclic N–C1 and Cα–Cε bonds (see Scheme 1B for the atom nomenclature), we provide the first detailed insights into the three-dimensional solution structures of these reactive intermediates in organocatalysis.s-cis and s-trans: Conformation of the exocyclic N–C1 bond
Shifting the equilibrium between the two enamine conformations s-cis and s-trans (with respect to the N–C1 single bond with partial double bond character, Fig. 1A) towards the s-trans conformation, most likely by steric repulsion, is one of the two proposed functions of the (diaryl)methanol (ether) substituent in the α–position of organocatalysts 1–7.39b In analogy to the relative s-cis and s-trans enamine stabilities, a preference of the corresponding isomeric E-iminium ion over the Z-isomer can be assumed, as both biases are thought to originate from the same steric effect (Fig. 1A).39b The guarantee of this basic conformational feature of the enamine key intermediate by the bulky α-substituent is believed to be essential for the stereochemical outcome of prolinol(ether)-catalyzed reactions. For instance, we have recently pointed out the stereochemical implication of the s-ciss-trans enamine equilibrium for the formation of the isomeric cyclic oxazolidines by prolinol catalysts.46 The predominance of the s-trans conformation in the case of proline enamines has been proposed from calculations48 and has been experimentally proven by our NMR studies in solution;47 in addition, exclusively s-trans proline enamines have been detected in crystal structures.49 Likewise for prolinol ether enamines, the s-trans conformation has been observed in crystal structures39 and its energetic preference has been thoroughly calculated.39,41,43–45 Interestingly, the generally accepted assumption that s-transenamines of diarylprolinol silyl ethers are a lot more stable than the s-cisenamines has recently been challenged by a theoretical study that predicted similar energies and, hence, similar populations of both conformations using gas-phase calculations.44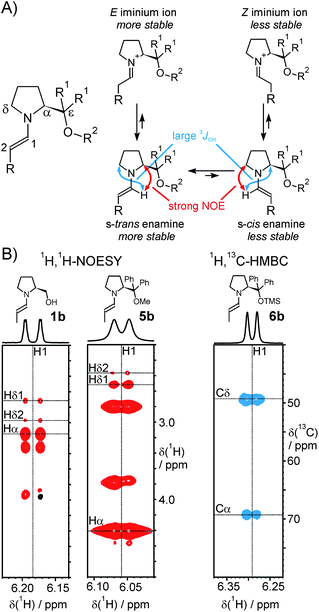 | ||
| Fig. 1 A) Left: atom nomenclature; right: the equilibrium between the enamine conformations, their relation to iminium ions and the distinctive NOEs and 3JCH; B) Sections of the 1H,1H-NOESY spectra of 1b (left) and 5b (middle) and of a 1H,13C-HMBC spectrum of 6b (right) in DMSO-d6 at 300 K. | ||
To experimentally clarify this issue in solution, we analyzed the 1H,1H-NOESY spectra of enamines 1b–7b (example sections are shown in Fig. 1B). The relative intensities of the NOEs between H1 and the protons Hα or Hδ1,2 are considered to be a suitable indicator for the differentiation between the s-trans and the s-cis conformations (Fig. 1A). For the s-trans conformation, a stronger NOE between H1 and Hα is expected, while a stronger NOE between H1 and Hδ1,2 would be indicative of a predominant population of the s-cis conformation. The 1H,13C-HMBC cross-peak intensities between H1 and Cα or Cδ can be used as an additional criterion, since 3JCH couplings are known to be larger in an antiperiplanar than in a synperiplanar arrangement.50 Accordingly, a larger HMBC cross-peak H1–Cδ is indicative of the s-trans conformation, whereas the s-cis conformation would be revealed by a larger H1–Cα cross-peak. In our NOESY experiments, significantly more intensive cross-peaks from H1 to Hα than to Hδ1,2 were observed for all of the enamines investigated, i.e. for 1b, 3a-b, 5a-b, 6a-b and 7b in DMSO-d6, for 5b in CDCl3 and for 7b in PhMe-d8. This indicates that the s-trans conformation is indeed preferably populated by enamines derived from α-substituted pyrrolidines and different aldehydes in both polar and non-polar solutions. This finding was confirmed by the 1H,13C-HMBC spectrum of enamine 6b. The more intensive cross-peak between H1 and Cδ in comparison to H1 and Cα (Fig. 1B, right) indicates a larger 3JHC coupling between H1 and Cδ and, thus, also reveals the preferred adoption of the s-trans conformation.
Furthermore, using quantitative NOESY analyses we studied to what extent the methanol (ether) substituent impacts on the actual position of the s-cis s-trans equilibrium (Table 1). For this purpose, the volume of the NOESY cross-peak between H1 and Hα was compared to the sum of the cross-peak volumes between H1 and Hδ1,2. The larger the ratio NOE(H1–Hα):NOEs(H1–Hδ1,2), the larger the contribution of the s-trans conformation to the s-cis s-trans equilibrium is in solution. The theoretical ratio NOE(H1–Hα):NOEs(H1–Hδ1,2) for a pure s-trans enamine was calculated on the basis of the internuclear distances from the DFT-optimized s-trans prolinol ether enamine structures provided in the literature.44,45 From this calculation, the maximum NOE ratio is about 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for the pure s-trans conformation and, accordingly, cannot be exceeded further (Table 1, right).
:1 for the pure s-trans conformation and, accordingly, cannot be exceeded further (Table 1, right).
|
|
Normalized NOESY cross-peak volumes | ||||
|---|---|---|---|---|---|
| Experimental values | Theoretical values (s-transenamines) | ||||
| NOE pair | 1b | 5b, 6b, 7b | 5a, 6a | 6b 45 | 7b 44 |
|
a The short lifetimes of diphenylprolinol enamines 3a and 3b, resulting in poor spectral resolution, did not allow a reliable NOESY integration.
b For 1b, the peaks of Hα and one of the protons Hε overlap. Therefore, the ratio of 8:2 is an upper limit and the actual increase in the cross-peak ratio from 1b to 5b/6b/7b should be significantly higher.
|
|||||
| R1 = H | R1 = Ar | R1 = Ph | R1 = Ph | R1 = Ar2 | |
| R2 = H | R2 = Me/TMS | R2 = Me/TMS | R2 = TMS | R2 = TMS | |
| R = Me | R = Me | R = iPr | R = Me | R = Me | |
| H1-Hα | ≤79b | 91–94 | 90–91 | 88 | 92 |
|
|||||
| H1–Hδ1 + H1–Hδ2 | ≥21b | 6–9 | 9–10 | 12 | 8 |
From the experimental NOESY cross-peak integration of enamine 1b (Table 1), it becomes obvious that the theoretical value for the pure s-trans enamine conformation (about 9:1) is not reached for prolinol (1). This indicates the partial adoption of the s-cis conformation by 1b and, hence, represents the first experimental evidence that the s-cis conformation significantly contributes to the conformational ensemble of a prolinol enamine in solution. This interpretation is also supported by the recently reported slow equilibration of the isomeric prolinol oxazolidines, presumably via the s-trans–s-cisisomerization of the enamine.46 In contrast, the increasing sizes of the pyrrolidine α-substituents in the catalysts 5–7 lead to an increase in the NOESY cross-peak volume ratio NOE(H1–Hα):NOEs(H1–Hδ1,2), from less than 8:2 to more than 9:1. This indicates that bulkier α-substituents indeed enforce the strong preference for the s-trans enamine conformation. However, interestingly, there is no additional visible increase in the NOESY cross-peak ratio with further enlargement of the pyrrolidine α-substituent (e.g. from 5b over 6b to 7b) or the aldehyde alkyl chain (compare entries for 5b,6b with 5a,6a). For all of these diarylprolinol ether enamines, the congruence of the experimental NOE ratios of about 9:1 with the theoretical values for the pure s-trans conformation suggests that the “saturation” of the NOESY cross-peak ratio and, accordingly, of the corresponding s-cis to s-trans population ratio can be understood in terms of an almost exclusive adoption of the s-trans conformation. In addition, this postulation of a negligible s-cis population of diarylprolinol ether enamines is also in line with our observations on the exclusive formation of the endo-oxazolidines by diarylprolinol catalysts.46
Conformation of the pyrrolidine ring: up and down
To the best of our knowledge, no attention has been paid to the potential influence of the puckering of the pyrrolidine ring on the overall conformation of enamines derived from prolinol-based organocatalysts. Only for proline-derivatives has the pyrrolidine conformation in aldol transition states been theoretically studied.51 This previous lack of interest is striking in view of the fact that the pyrrolidine ring is known to be an important structure,27,52–54 since proline as an organocatalyst has been found to provide significantly better yields and stereoselectivities than related catalysts with four- or six-membered rings. Accordingly, in the context of diarylprolinol ether enamines, only one single comment on the pyrrolidine conformation has become known to us from an X-ray study that states that “the puckering of the pyrrolidine ring varies from structure to structure”.39 However, to our mind, the conformational preferences of the pyrrolidine ring should be considered in more detail for two reasons. Firstly, pyrrolidine hydrogen atoms may potentially participate in stabilizing CH/π interactions55–57 with the phenyl rings in diarylprolinol (ether) enamines; these weak interactions have been proven to have important implications not only in biochemistry,58,59 but also in molecular recognition and organic chemistry.60–63 Secondly, in general, different pyrrolidine ring conformations may well be associated with different reactivities64 and the catalytic performances of the respective compounds.65 In particular, in diarylprolinol ether enamines the pyrrolidineup conformation, in combination with the known slight pyramidality of the enamine nitrogen atom,39–41,43–45,51 creates a concave surface for the attack of the electrophile (the convex surface is supposed to be shielded by the “obese” α-substituent, Fig. 2A, left), which is known to be a sterically unfavorable situation. In contrast, in the down conformation the enamine surface opposite to the “obese” substituent is convex and, hence, is wide-open for the electrophilic attack towards the enamine. | ||
| Fig. 2 A) The two low-energy pyrrolidine conformations “up” and “down”. B) Distinguishing the calculated 3JHH coupling constants for the up and down conformations of proline residues in proteins66 and the experimental range for the enamines investigated in this study. C) Sections of the 1D 1H NMR spectrum of 6a in DMSO-d6 at 300 K showing the typical multiplet patterns for Hα, Hδ1 and Hδ2 observed in diarylprolinol (ether) enamines. | ||
As a basis for studying the pyrrolidine conformation in prolinol (ether) enamines, the previous extensive investigations on proline side-chain conformations could be used. For proline residues in peptides and proteins, it has been established that two distinct pyrrolidine envelope conformations are preferentially adopted, commonly designated as “up” and “down” (Fig. 2A). This simple two-state model for the pyrrolidine ring in proline should be readily transferable to prolinol (ether) enamines, since approximate planarity may be assumed for both the amide group in peptides and the enamine moiety in organocatalytically active intermediates (on the basis of prolinol ether enamine crystal structures39 and DFT calculations39b,41,43–45). In addition, the scalar coupling constant (J) criteria for proline side-chain conformations can be applied also to diarylprolinol (ether) enamines, as no systematic shift of the 3J(Hα,Hβ1/2), potentially caused by the different Cε-substituents, is observed for the free catalysts proline and 2–7 (see Schemes S3 and S4 in the supplementary information†). The two different pyrrolidine conformations up/down can be distinguished by NMRvia their characteristic 3JHH,66 which are easily extracted from well-resolved 1H resonance multiplet patterns. Accordingly, small 3J(Hα,Hβ2) and 3J(Hδ2,Hγ1) indicate the population of the down conformation, while small 3J(Hδ1,Hγ2) are indicative of the up conformation. The two conformations up/down and the associated theoretical and experimentally observed 3JHH values are summarized in Fig. 2.
For diarylprolinol (ether) enamines 2a–7b, small vicinal couplings of 1.5–2.5 Hz and 2–3 Hz, respectively, were found for 3J(Hα,Hβ2) and 3J(Hδ2,Hγ1), (not only in DMSO-d6, but also in MeCN-d3, CDCl3 and PhMe-d8, see Scheme S1 in the supplementary information†). In contrast, values in the range 6–10 Hz were detected for 3J(Hδ1,Hγ2), which leads to the characteristic multiplet patterns depicted in Fig. 2C for the example of 6a. (Unfortunately, coupling constants could not be extracted for 1a and 1b due to spectral overlap and higher order NMR signals.) The experimental values for 3J(Hα,Hβ2) and 3J(Hδ2,Hγ1) equal those expected for the pure down conformation.66 This indicates the down conformation for the pyrrolidine ring in diarylprolinol (ether) enamines in solvents ranging from DMSO over MeCN to CHCl3 and PhMe. In addition, the small 3J(Hα,Hβ2) and 3J(Hδ2,Hγ1) show that conformations with large coupling constants, e.g. up, do not substantially contribute to the conformational ensemble, which can be taken as an indication of a rather stable structure.67 Interestingly, for the aldol transition states of the proline-derived catalysts, the theoretical calculations suggested that the down conformation is significantly preferred only for β-substituted pyrrolidine rings.51 However, our experimental study reveals a high preference for the down conformation, even in the absence of β-substituents. In contrast, in free catalysts 2–7, both 3J(Hα,Hβ1/2) are larger than 7 Hz, which indicates a dynamic equilibrium of the up and down conformations (see Schemes S3 and S4† and the exemplary Hα multiplets in Fig. 3C of our previous report.46).
 | ||
| Fig. 3 A) Staggered conformations of diarylprolinol (ether) enamines. B) 1H NMR assignments of 3b (left) and 5b (right) (note: the chemical shifts of Hβ1, Hγ1 and Hδ1 are listed below those of Hβ2, Hγ2 and Hδ2) and sections of their 1H,1H-NOESY spectra (bottom) in DMSO-d6 at 300 K (intensities of the sections are scaled individually for optimum clarity). | ||
These experimental results show that the enamine formation is essential for the adoption of a conformational preference of the pyrrolidine ring. This means that the approximate planarity of the enamine moiety, along with the bulky α-substituent, imposes conformational constraints on the pyrrolidine ring to such a degree that one pyrrolidine conformation (down) is exclusively observed. As a first assumption, this may be rationalized by the different steric repulsion modes within the up and down conformations presented in Fig. 2A. The up conformation may be destabilized by the detrimental repulsion between the “obese” α-substituent and the vicinal β, as well as the δ protons, which is reduced in the preferred down conformation (Fig. 2A; this hypothesis parallels the observed slight pyramidalization of the enamine nitrogen known from crystal structure analyses39 and DFT calculations.39b,41,43–45,51) Furthermore, for proline derivatives, the down conformation has been calculated to be compatible with less deviation of the enamine moiety from the favorable planarity than the up conformation.51 In addition, it is only the down conformation that creates sufficient spatial proximity between the methanol ether substituents and the Hγ2 of the pyrrolidine ring (Fig. 2A) to potentially allow for stabilizing CH/π interactions (see below). Finally, only in the down conformation, the attack of an electrophile occurs in a sterically favorable manner to the unshielded and convex surface of the enamine.
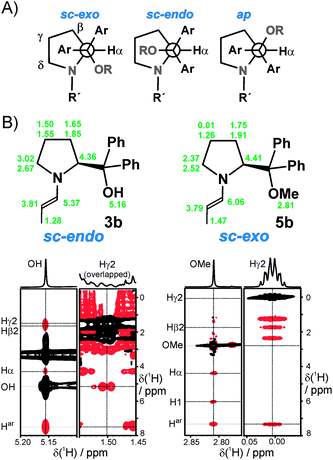
Orientation of the diarylmethanol substituent by rotation around the exocyclic Cα–Cε bond: sc-exo, sc-endo and ap
The effective shielding of one face of the enamine π system, leading to the approach of incoming electrophiles from the opposite side, is meant to be the second important function of the diaryl-methanol (ether) substituent for the stereochemical outcome of enamine-catalyzed reactions by organocatalysts 2–7.39b However, beyond empirical experience on catalyst performances, very little is known about whether this shielding is brought about by the O-protecting group or the phenyl rings of the “obese” substituents of diarylprolinol(ether)-type organocatalysts. This issue, which is highly important for theoretical calculations aimed at the understanding of the stereoselection, is closely connected to the conformation of the exocyclic Cα–Cε bond (see Scheme 1B). Rotation around this bond is supposed to be rather fixed by the geminal-diaryl effect.39a Again, the available conformational information has been limited to crystal structure analyses39a and to theoretical calculations.39b,41,43–45 However, because of the lack of experimental data in solution, partially conflicting results have been put forward, in particular whether the sc-exo39b,45 or the sc-endo41,43,44 conformation of diarylprolinol ether enamines constitutes the better structural basis for intermediate and transition state calculations. This also holds true for (diaryl)prolinol-derived enamines, for which two opposite modes of stereoselection have been claimed: steric shielding of one face of the enamine by the aryl rings36 on the one hand and direction of the electrophile to this face of the enaminevia an H-bond35,37 on the other hand. Knowledge of the rotation around the Cα–Cε bond should also help to shed some light on this issue.The spectral sections of 3b reveal significantly stronger NOEs from OH to Hβ2 and Hγ2 than to Hα and a stronger NOE of Hγ2 to OH than to the aromatic protons of the phenyl rings (Fig. 3B, left). This NOE pattern is best explained by an sc-endo conformation of the Cα–Cε bond in the case of 3b. In contrast, for 5b, the protons of OMe show much stronger NOEs to H1 and Hα than to Hβ2 or Hγ2 and, vice versa, a stronger NOE from Hγ2 to the aromatic protons than to the protons of OMe was observed (Fig. 3B, right). These findings for 5b are indicative of an sc-exo conformation around the exocyclic Cα–Cε bond. In line with the gauche effect, the preferential adoption of the ap conformation, however, can be ruled out on the basis of these NOE intensity patterns in both cases.
 | ||
| Fig. 4 A) 1H chemical shifts of 3b, 1b, and 5b in DMSO-d6 with upfield-shifted resonances highlighted in red (left) and sections of the corresponding 1H NMR spectra (right). (Hβ1, Hγ1 and Hδ1 are listed below Hβ2, Hγ2 and Hδ2. For 1b, the Cα–Cε conformation is not accessible because of signal overlap.) B) MMFF-refined structure models of 3b and 5b based on the NMR-derived conformational features. | ||
In contrast to all of the enamines studied, neither the conformational fixation of the pyrrolidine ring (see above) nor the upfield-shifts of individual protons are observed for the free catalysts 2–7 (see Schemes S3 and S4 in the Supplementary Information†). One may thus assume that the predictive value of conformational studies on prolinol (ether) organocatalysts for the conformations of their enamine intermediates is rather limited. Instead, our investigations stress the importance of performing conformational studies on the actual organocatalytic intermediates themselves as reliable starting points for theoretical calculations of the reaction pathways and transition state conformations (see discussion below). In addition, the simultaneous appearance of the conformational preferences of the pyrrolidine ring and around the Cα–Cε bond in prolinol (ether) enamine intermediates strongly suggest stabilizing interactions between the pyrrolidine ring and the methanol ether substituents (in agreement with the interactions discussed above and shown in Fig. 4).
On the basis of their excellent correspondence with the sc-endo or sc-exo conformation around the exocyclic Cα–Cε bond, the upfield-shifts of protons H1 or Hγ2 and Hδ2, respectively, in the enamine intermediates can be used as a facile method to rapidly screen diarylprolinol-derived enamines for the orientation of the bulky diarylmethanol substituent. As the case of the ap conformation can be ruled out as the major conformation, as revealed for all of the enamines studied (see below), the chemical shifts for H1 of 5.20–5.42 ppm are indicative of the sc-endo conformation, while Hγ2 and Hδ2 resonances in the ranges 0.00–0.35 ppm and 2.20–2.40 ppm, respectively, evidence the sc-exo conformation.
| Enamine | δ (1H)/ppm | NOESY-based conformation | ||
|---|---|---|---|---|
| H1 | Hγ2 | Hδ2 | ||
| a Only the chemical shift ranges can be given because of severe resonance overlap. b Spectral overlap prevented the determination of the conformation. c n. ass. = not assignable; n. det. = not determined. | ||||
| 1a | 6.17 | 1.75–1.45a | 2.97 | n. ass.b |
| 1b | 6.18 | 1.76 | 2.98 | n. ass.b |
| 2a | 5.27 | 1.80–1.35a | 3.07 | n. det. |
| 2b | 5.42 | 1.65–1.35a | 3.04 | n.det. |
| 3a | 5.26 | 1.55 | 3.06 | sc-endo |
| 3b | 5.37 | 1.50 | 3.02 | sc-endo |
| 4a | 5.20 | 1.75–1.30a | 3.11 | n. det. |
| 4b | 5.37 | 1.65–1.35a | 3.09 | n. det. |
| 5a | 5.94 | 0.01 | 2.37 | sc-exo |
| 5b | 6.06 | 0.01 | 2.37 | sc-exo |
| 6a | 6.19 | 0.34 | 2.33 | sc-exo |
| 6b | 6.29 | 0.25 | 2.34 | sc-exo |
| 7a | 6.05 | 0.30 | 2.23 | n. det. |
| 7b | 6.22 | 0.25 | 2.27 | sc-exo |
| Enamine | Solvent | δ (1H)/ppm | NOESY-based conformation | ||
|---|---|---|---|---|---|
| H1 | Hγ2 | Hδ2 | |||
| a n. det. = not determined. | |||||
| 5b | DMSO-d6 | 6.06 | 0.01 | 2.37 | sc-exo |
| MeCN-d3 | 6.14 | 0.08 | 2.43 | n. det. | |
| MeOH-d4 | 6.10 | 0.14 | 2.44 | n. det. | |
| CDCl3 | 6.11 | 0.10 | 2.48 | sc-exo | |
| PhMe-d8 | 6.31 | 0.22 | 2.53 | n. det. | |
| 7b | DMSO-d6 | 6.22 | 0.25 | 2.27 | sc-exo |
| PhMe-d8 | 6.12 | 0.09 | 2.26 | n. det. | |
We first examined the potential influences of the catalyst structure and the aldehyde alkyl chain on the Cα–Cε conformation (Table 2). By comparison to the ring current-free enamines 1a and 1b, upfield shifts of the H1-resonance in all of the O-unprotected enamines 2a–4b become evident, as well as upfield shifts of the Hγ2/Hδ2-resonances of all of the O-protected enamines 5a–7b (entries in Table 2 highlighted in grey). As verified in most cases by NOESY analyses, these shifts are indicative of the sc-endo conformation for all of the diarylprolinol enamines (2a–4b) and of the sc-exo conformation for all of the diarylprolinol ether enamines (5a–7b).
In addition, the possible solvent effects on the preferred population of these conformations were investigated (Table 3). As the detection ofprolinol enamines was only successful in DMSO-d6, these solvent studies were performed for only the diarylprolinol ether enamines, on the examples of 5b and 7b. The characteristic upfield-shifts of Hγ2/δ2 were found in all of the solvents applied, ranging from polar aprotic (DMSO-d6, MeCN-d3) over polar protic (MeOH-d4) to nonpolar (CDCl3) and aromatic solvents (PhMe-d8). This indicates that solvent properties do not affect the conformational preferences around the Cα–Cε bond of diarylprolinol ether enamines.
Altogether, our straightforward 1H NMR screening method, backed by NOESY analyses, shows that the protection of the hydroxylic group is the decisive factor for the conformational switch observed from diarylprolinol enamines (sc-endo) to diarylprolinol ether enamines (sc-exo). In contrast, neither the nature of the protecting group (Me or TMS, cf.5a,b with 6a,b), nor the nature of the aromatic rings (Ph or Ar, cf.3a,b with 2a,b and 4a,b or cf.6a,b and 7a,b), nor the size of the aldehyde alkyl chain (iPr or Me, cf.2a–7a with 2b–7b) seem to be of greater conformational importance. Moreover, the sc-exo conformation is preferred by diarylprolinol ethers independent of the solvent used. Thus, from a conformational point of view, the etherification of the hydroxylic group of prolinols does not only have a significant impact on the stability of the corresponding enamines,46 but also on the orientation of the bulky pyrrolidine α-substituent.
 | ||
| Scheme 3 1H chemical shifts of diarylprolinol silyl ether enamines reported in the literature: 8 was observed in situ by Jørgensen et al.41 and 9 and 10 were isolated and investigated in Seebach's group.39b (Note: H1 of 9 and 10 had obviously been mis-assigned by accident in the literature.) | ||
Furthermore, our results provide the first broad experimental basis to clarify the recently presented conflicting results from theoretical calculations on the Cα–Cε conformation of diarylprolinol ether enamines. They clearly evidence the sc-exo conformation of s-trans diarylprolinol ether enamines in solution and thus, in agreement with a comparative theoretical study from Seebach's group,39b back the sc-exo conformation39b,45 and reject the sc-endo conformation41,43,44 as the proper basis for enamine intermediate calculations.
Beyond the determination of intermediate conformations, we believe our study is also relevant to the calculation and investigation of organocatalytic reaction pathways. In addition, our study allows the identification of theoretical studies that are in agreement with the structural properties, being valid in solution. For instance, a recent DFT calculation on the transition state for the asymmetric Michael addition of 5b to methyl vinyl ketone72 features all of the conformational properties of the enamine intermediate that we determined experimentally; the E-configuration of the enamine double-bond, the s-trans arrangement of the enamine, the down conformation of the pyrrolidine ring and the sc-exo conformation of the α-substituent around the Cα–Cε bond. Accordingly, the electrophilic attack of methyl vinyl ketone to the enamine occurs from the convex half-space opposite the “obese” diphenylmethoxymethyl substituent of 5b. As previously pointed out,39 in the sc-exo conformation of diarylprolinol ether enamine intermediates, the steric shielding of one face of the enamine is secured by both the meta-substituents of the aryl groups and the O-protecting group. Thus, increasing stereoselectivities in asymmetric reactions should be obtained by enlarging either the aryl meta-substituent or the O-protecting group of the organocatalyst. Indeed, this effect has been regularly reported for increasing sizes of the aryl meta-substituent29,73–75 and the O-protecting group.28,75–78 In addition, our finding of a stable sc-exoCα–Cε conformation predicts that the enlargement of only one of the two phenyl rings should be sufficient to increase the shielding of one face of the enamine and hence to increase the stereoselectivity. In fact, such an effect has been recently observed.79 All this data suggests that the sc-exo45 conformation and not the sc-endo43–45 conformation is also predominant in transition states involving diarylprolinol ether enamines, which may be confirmed by further theoretical calculations based on this experimental study.
Our first experimental data on prolinol enamine intermediate conformations might be a useful guide for further theoretical investigations on the origin of stereocontrol by diarylprolinol enamines, despite their rather limited applicability. Still, it is interesting to note that for prolinol enamines two different modes of stereocontrol in the bond-forming transition state have been postulated. On the one hand, steric shielding of the “upper” enamine face by the bulky substituent has been proposed36 and, on the other hand, a directing function of the hydroxylic groupvia H-bonding interactions35,37,38,79 to the electrophile on this “upper” face has been claimed. Interestingly, the sc-endo conformation around the Cα–Cε bond of diarylprolinol enamines that we observed in this study allows for both a H-bond from the hydroxylic group to an incoming electrophile and steric shielding by one of the aryl rings. Thus, both interactions may indeed contribute as stereodirecting factors. Nevertheless, since the change of the sc-endo conformation in diarylprolinol enamines to sc-exo in diarylprolinol ether enamines is apparently triggered by the protection of the OH-functionality, a special role in the stabilization of the sc-endo conformation can be attributed to the OH group. This is in agreement with a previous study39 that shows that prolinol enamines may develop an N⋯HO hydrogen bond only in the sc-endo conformation (note: N is to be taken as a representative of the enamine π system as a hydrogen bond acceptor). In the case of our simple structure model of Fig. 4B (d (N⋯H) ≈ 2.1 Å, d (N⋯O) ≈ 2.7 Å, < (N⋯H–O) ≈ 122°), this hydrogen bond in 3b is to be classified as weak to moderate,80 but it might be sufficient to cause the preference of the sc-endo conformation. In solvents with lower H-bond acceptor abilities than DMSO, the favourable energetic contribution of this H-bond should be even more pronounced. Moreover, the upfield-shift of the H1 resonance (see above) indicates an additional CH/π contribution between H1 and one of the phenyl rings that also stabilizes the sc-endo conformation of diarylprolinol derivatives (Fig. 4B, top). For the further rationalization of the sc-endo conformation, the stronger steric repulsion between the pyrrolidine hydrogens and the aryl rings compared to the OH-group has been claimed previously.39 This would imply that the sc-exo conformation in diarylprolinol ether enamines should be switchable to sc-endo either by reducing the size of the O-protecting group (Me instead of TMS) or by increasing the size of the aromatic rings (Ar instead of Ph); yet, in none of these cases did we observe a change of the preferred sc-exo conformation towards sc-endo. This makes us believe that steric clashes are of minor importance for the issue of conformational preferences around the Cα–Cε bond. Thus, it is highly likely that the weak conformation-stabilizing intramolecular interactions account for the observed preferences of the sc-endo and the sc-exo conformations of diarylprolinol (ether) enamines. For diarylprolinol enamines, we found evidence for a N⋯HO hydrogen bond and one CH/π interaction and in diarylprolinol ether enamines strong experimental evidence for two CH/π interactions is provided. Thus, for the first time, CH/π interactions are suggested as a conformation-determining factor for enamine intermediates in organocatalysis. It is noteable that upfield-shifted pyrrolidine protons have also been reported for diarylprolinol ether iminium salts,39b,42 which, in combination with the crystallographic data,39 may be interpreted in terms of similar CH/π interactions being operative and structure-determining in iminium ions too.
Graphical summary of the conformational preferences and NMR screening methods
The crucial conformational aspects of diarylprolinol (ether) enamines (orientation of the ene moiety, pyrrolidine puckering and rotation of the bulky diarylmethanol substituent) can be straightforwardly screened for by means of NMR spectroscopy. For the sake of clarity, the results and approaches outlined above are summarized graphically in Fig. 5. | ||
| Fig. 5 A graphical summary of the conformational preferences and NMR screening methods for prolinol (ether) enamines. | ||
Conclusions
In summary, we present the first detailed conformational investigations on enamines derived from prolinol and prolinol ether-type organocatalysts, with two different aldehydes in various solvents, by means of NMR spectroscopy. Concerning the exocyclic N–C bond, we report the first NOESY-based experimental proof that a prolinol-derived enamine partially exists in the s-cis conformation in solution. For diarylprolinol ether enamines in contrast, only the s-trans conformation is observed in solution most probably owing to the bulkiness of the pyrrolidine α-substituent. In addition, for all of the enamines studied, enamine formation is associated with a strong preference for the down conformation of the pyrrolidine ring. For the rotation around the exocyclic Cα–Cε bond, diarylprolinol enamines are found by NOESY analyses to be present in the sc-endo conformation, while the diarylprolinol ether enamines adopt the sc-exo conformation. Strong experimental evidence is provided that the sc-exo conformation in diarylprolinol ether enamines is stabilized by CH/π interactions between the aliphatic hydrogen atoms of the pyrrolidine ring in the down conformation and an aromatic π system of the bulky pyrrolidine α-substituent. In addition, a rapid conformational screening method, based on 1H chemical shifts, was developed and applied to show the generality of these conformational preferences for various catalysts, aldehydes and solvents.The broad experimental basis provided in this study and our observation of the exquisite conformational preferences of enamine intermediates in solution experimentally clarify the hitherto contradictory postulations and unsolved issues of s-cis/s-trans and sc-endo/sc-exoenamine conformations. Thus, the presented conformational features help to explain the experimental performances of various catalysts, promote the rationalization of the stereochemical outcome and facilitate further catalyst optimization.
Acknowledgements
This work was supported by the DFG (SPP 1179). Scholarships from the Cusanuswerk and the Studienstiftung des deutschen Volkes are gratefully acknowledged.Notes and references
- A. Berkessel and H. Gröger, Asymmetric Organocatalysis—From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- Enantioselective Organocatalysis: Reactions and Experimental Procedures, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Special Issue on Organocatalysis, Chem. Rev., 2007, Search PubMed 107, 12.
- Ernst Schering Foundation Symposium Proceedings “Organocatalysis”, ed. M. T. Reetz, B. List, S. Jaroch and H. Weinmann, Springer, Berlin, 2008 Search PubMed.
- Asymmetric Organocatalysis, ed. B. List , 2010 Search PubMed.
- J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719–724 CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS.
- T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656–5682 CrossRef CAS.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS.
- P. M. Pihko, ed., Hydrogen Bonding in Organic Synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed.
- K. Etzenbach-Effers and A. Berkessel, Top. Curr. Chem., 2010, 291, 1–27 CrossRef CAS.
- T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS.
- D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395–456 CrossRef CAS.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS.
- B. List, Chem. Commun., 2006, 819–824 RSC.
- P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS.
- S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS.
- P. M. Pihko, I. Majander and A. Erkkilä, Top. Curr. Chem., 2010, 291, 29–75 CrossRef CAS.
- A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef.
- J. B. Brazier and N. C. O. Tomkinson, Top. Curr. Chem., 2010, 291, 281–347 CrossRef CAS.
- T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS.
- J. J. Devery III, J. C. Conrad, D. W. C. MacMillan and R. A. Flowers II, Angew. Chem., Int. Ed., 2010, 49, 6106–6110 CrossRef.
- J. M. Um, O. Gutierrez, F. Schoenebeck, K. N. Houk and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 6001–6005 CrossRef CAS.
- U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- Y. Chi and S. H. Gellman, Org. Lett., 2005, 7, 4253–4256 CrossRef CAS.
- M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS.
- Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS.
- C. Palomo and A. Mielgo, Angew. Chem., Int. Ed., 2006, 45, 7876–7880 CrossRef CAS.
- A. Mielgo and C. Palomo, Chem.–Asian J., 2008, 3, 922–948 CrossRef CAS.
- L. W. Xu, L. Li and Z. H. Shi, Adv. Synth. Catal., 2010, 352, 243–279 CrossRef CAS.
- A. Lattanzi, Chem. Commun., 2009, 1452–1463 RSC.
- G. Zhong, J. Fan and C. F. Barbas III, Tetrahedron Lett., 2004, 45, 5681–5684 CrossRef CAS.
- D. Enders and S. Chow, Eur. J. Org. Chem., 2006, 4578–4584 CrossRef CAS.
- Y. Hayashi, T. Itoh, S. Aratake and H. Ishikawa, Angew. Chem., Int. Ed., 2008, 47, 2082–2084 CrossRef CAS.
- Y. Hayashi, S. Samanta, T. Itoh and H. Ishikawa, Org. Lett., 2008, 10, 5581–5583 CrossRef CAS.
- (a) D. Seebach, U. Grošelj, D. M. Badine, W. B. Schweizer and A. K. Beck, Helv. Chim. Acta, 2008, 91, 1999–2034 CrossRef CAS; (b) U. Grošelj, D. Seebach, D. M. Badine, W. B. Schweizer, A. K. Beck, I. Krossing, P. Klose, Y. Hayashi and T. Uchimaru, Helv. Chim. Acta, 2009, 92, 1225–1259 CrossRef.
- For a recent report on an O-methyl prolinol derived enamines, see: P. Domínguez de María, P. Bracco, L. Fernando Castelhano and G. Bargeman, ACS Catal., 2011, 1, 70–75 CrossRef.
- S. Bertelsen, M. Marigo, S. Brandes, P. Diner and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973–12980 CrossRef CAS.
- S. Lakhdar, T. Tokuyasu and H. Mayr, Angew. Chem., Int. Ed., 2008, 47, 8723–8726 CrossRef CAS.
- J. Franzén, M. Marigo, D. Fielenbach, T. C. Wabnitz and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 18296–18304 CrossRef.
- P. Dinér, A. Kjærsgaard, M. A. Lie and K. A. Jørgensen, Chem.–Eur. J., 2008, 14, 122–127 CrossRef.
- J.-Q. Zhao and L.-H. Gan, Eur. J. Org. Chem., 2009, 2661–2665 CrossRef CAS.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, J. Am. Chem. Soc., 2011, 133, 7065–7074 CrossRef CAS.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, Angew. Chem., Int. Ed., 2010, 49, 4997–5003 CrossRef CAS.
- S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475–2479 CrossRef CAS.
- D. A. Bock, C. W. Lehmann and B. List, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20636–20641 CrossRef CAS.
- M. Karplus, J. Chem. Phys., 1959, 30, 11–15 CrossRef CAS.
- C. Allemann, J. M. Um and K. N. Houk, J. Mol. Catal. A: Chem., 2010, 324, 31–38 CrossRef CAS.
- J. Ammer, M. Baidya, S. Kobayashi and H. Mayr, J. Phys. Org. Chem., 2010, 23, 1029–1035 CrossRef CAS.
- T. Kanzian, T. A. Nigst, A. Maier, S. Pichl and H. Mayr, Eur. J. Org. Chem., 2009, 6379–6385 CrossRef CAS.
- G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz and R. Terrell, J. Am. Chem. Soc., 1963, 85, 207–222 CrossRef CAS.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction: Evidence, Nature, and Consequences, Wiley, New York, 1998 Search PubMed.
- S. Tsuzuki and A. Fujii, Phys. Chem. Chem. Phys., 2008, 10, 2584–2594 RSC.
- M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC.
- M. Brandl, M. S. Weiss, A. Jabs, J. Sühnel and R. Hilgenfeld, J. Mol. Biol., 2001, 307, 357–377 CrossRef CAS.
- G. Toth, S. G. Bowers, A. P. Truong and G. Probst, Curr. Pharm. Des., 2007, 13, 3476–3493 CrossRef CAS.
- M. Nishio and M. Hirota, Tetrahedron, 1989, 45, 7201–7245 CrossRef CAS.
- M. Nishio, Y. Umezawa, M. Hirota and Y. Takeuchi, Tetrahedron, 1995, 51, 8665–8701 CrossRef CAS.
- M. Nishio, Tetrahedron, 2005, 61, 6923–6950 CrossRef CAS.
- G. Tárkányi, P. Király, S. Varga, B. Vakulya and T. Soós, Chem.–Eur. J., 2008, 14, 6078–6086 CrossRef.
- L.-S. Sonntag, S. Schweizer, C. Ochsenfeld and H. Wennemers, J. Am. Chem. Soc., 2006, 128, 14697–14703 CrossRef CAS.
- D. A. DiRocco, K. M. Oberg, D. M. Dalton and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10872–10874 CrossRef CAS.
- M. Cai, Y. Huang, J. Liu and R. Krishnamoorthi, J. Biomol. NMR, 1995, 6, 123–128 CrossRef CAS.
- Z. L. Mádi, C. Griesinger and R. R. Ernst, J. Am. Chem. Soc., 1990, 112, 2908–2914 CrossRef.
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of Organic Compounds, Wiley, New York, 1994 Search PubMed.
- C. Sparr, W. B. Schweizer, M. HansSenn and R. Gilmour, Angew. Chem., Int. Ed., 2009, 48, 3065–3068 CrossRef CAS.
- L. W. Reeves and W. G. Schneider, Can. J. Chem., 1957, 35, 251–261 CrossRef CAS.
- C. E. Johnson Jr. and F. A. Bovey, J. Chem. Phys., 1958, 29, 1012–1014 CrossRef.
- M. P. Patil, A. K. Sharma and R. B. Sunoj, J. Org. Chem., 2010, 75, 7310–7321 CrossRef CAS.
- M. Tiecco, A. Carlone, S. Sternativo, F. Marini, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2007, 46, 6882–6885 CrossRef CAS.
- Q. Zhu and Y. Lu, Org. Lett., 2008, 10, 4803–4806 CrossRef CAS.
- E. Marqués-López, R. P. Herrera, T. Marks, W. C. Jacobs, D. Könning, R. M. de Figueiredo and M. Christmann, Org. Lett., 2009, 11, 4116–4119 CrossRef.
- Y. Chi and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 6804–6805 CrossRef CAS.
- H. Gotoh and Y. Hayashi, Chem. Commun., 2009, 3083–3085 RSC.
- I. Mager and K. Zeitler, Org. Lett., 2010, 12, 1480–1483 CrossRef CAS.
- R. M. de Figueiredo, R. Fröhlich and M. Christmann, Angew. Chem., Int. Ed., 2008, 47, 1450–1453 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectroscopic characterization data of the organocatalysts and enamines. See DOI: 10.1039/c1sc00274k |
| This journal is © The Royal Society of Chemistry 2011 |