DOI:
10.1039/C1SC00203A
(Edge Article)
Chem. Sci., 2011,
2, 1788-1792
An anti-cancer trinuclear ruthenium(III) complex with 2-thiosalicylate ligands attenuates Wnt-β-catenin signaling†
Received
31st March 2011
, Accepted 27th May 2011
First published on 7th July 2011
Abstract
A new trinuclear ruthenium complex [RuIII3(TSA-H)2(TSA)4][NEt4] (1) with the non-toxic 2-thiosalicylic acid (TSA-H2) ligand could be prepared in a pure form and in a large quantity by a one-pot reaction. Complex 1 is stable under physiological conditions and forms adduct(s) with glutathione (GSH). It is anti-cancer active towards a panel of cancer cell lines, but does not exert its cytotoxicity through DNA binding. This is the first ruthenium complex reported to significantly attenuate the Wnt-β-catenin signaling in both transcriptomic and proteomic levels as demonstrated by DNA microarray, RT-PCR and Western blotting analyses.
Introduction
Identification of new anti-cancer metal complexes is presently receiving a surge of interest for their structural diversity and inimitable biological and medicinal properties arising from either covalent or non-covalent bindings/interactions with biomolecular cellular targets.1,2 Metal–ligand coordination is a useful means to create bioactive molecules with structural complexity and functionalities by one-pot reactions. In these endeavors, mononuclear metal complexes, such as that of platinum,3gold,4 and ruthenium5–8 have been documented to display promising anti-cancer activities. Meggers and co-workers have designed rigid octahedral ruthenium(II) and iridium(III) complexes to target the active site of large kinases.2 Two of the anti-cancer active ruthenium complexes, (H2im)[trans-RuCl4(Him)(DMSO)] (NAMI-A, wherein Him = imidazole)5 and (H2ind)[trans-RuCl4(Hind)2] (KP1019, wherein Hind = indazole),6 have entered clinical trials. A series of ruthenium(II)-arene complexes were found to display convincing cytotoxic and anti-metastatic activities.7 Recent works also revealed that ruthenium(III) complexes with pyrazolato and/or pyrazole ligands display dual anti-angiogenic and cytotoxic activities.8
Polynuclear metal complexes are known to display intriguing electronic structures and useful catalytic activities, and could be used as building blocks for the construction of molecular materials.9 In contrast, studies on the medicinal properties of polynuclear metal complexes are conspicuously sparse.10–12 The notable examples include dinuclear rhodium(II) carboxylates which display prominent in vitro and in vivo activities by inhibiting nucleic acid and protein synthesis.11 Polynuclear metal complexes are envisaged to have intriguing biological activities for the following reasons: (i) metal–metal bonded complexes may offer a new dimension in the construction of more complicated and sizable bioactive scaffolds with hydrophobicity for non-covalent binding with biomolecular cellular targets having large binding pocket; (ii) if the bridging ligands are non-toxic, polynuclear metal complexes could act as pro-drugs where a higher concentration of bioactive metal ions is feasible to be delivered to the cellular targets under in vivo conditions. Prominent examples are the polynuclear platinum(II) complexes12 such as [{trans-PtIICl(NH3)2}2μ-{trans-Pt(NH3)2(H2N(CH2)6NH2)2}]4+ and the polynucelar rhodium(II) butyrate complexes11 which have entered clinical trials. These complexes have been shown to display additional advantages over their mononuclear counterparts by offering higher biological efficacies and better kinetic stability.
The relationship between high activity on Wnt signaling and the occurance (etiology) of various types of cancers including liver and breast carcinomas has recently been uncovered.13 Several organic non-steroidal anti-inflammatory drugs (NSAID) such as indomethacin, sulindac and aspirin which entered clinical trials, have been identified as inhibitors of Wnt-signaling by repression of β-catenin expression and/or decreased expression of Wnt targeted genes.14 Contrary to these inhibitors, several ruthenium complexes with pyridocarbazole ligands were reported to significantly inactivate glycogen synthase kinase 3 (GSK-3),2 resulting in the positive regulation on Wnt signaling.
The captioned trinuclear ruthenium complex [RuIII3(TSA-H)2(TSA)4][NEt4] (1) with non-toxic 2-thiosalicylic acid (TSA-H2) bridging ligand could be easily prepared in a pure form and in a large quantity. It is relatively stable under physiological conditions and forms adduct with glutathione (GSH), a biological reducing tripeptide which is highly overexpressed in various types of cancer cells.15Notably, this anti-cancer active ruthenium complex shows inhibitory activities on Wnt-β-cateninsignaling pathway, which has scarcely been reported for anti-cancer metal-based drugs.4a
Results and discussion
Synthesis and characterization
The 2-thiosalicylic acid ligand offers several advantages for the construction of bioactive ruthenium complexes including that the ligands themselves are relatively non-toxic, and also the ease in the formation of multi-nuclear and/or multi-charged species. Solvothermal reaction was used to prepare the ruthenium-TSA complex. We found that solvothermal reaction of Ru(acac)3 (acac− = 2,4-pentanedionato) with TSA-H2 in the presence of NEt4OAc afforded [RuIII3(TSA-H)2(TSA)4][NEt4] (1) in 45% yield (complex 1, Fig. 1). Dark-purple crystals were obtained after solvent evaporation. Variable-temperature magnetic susceptibility measurement revealed that the complex has a magnetic state with S= ½ (Fig. S1, Electronic Supporting Information, ESI†). Its structure has been determined by X-ray crystallography (Fig. 2, CCDC-804342). The asymmetric unit of 1 contains two kinds of symmetry-inequivalent Ru atoms (two Ru1 and one Ru2) and three TSA ligands. Each TSA ligand binds two Ru atoms via one μ-S bridging and one S,O-chelating mode, leading to a linear S-bridged face-sharing tri-octahedral (FSTO) [Ru3] unit. The Ru⋯Ru distance of 2.736(1) Å is slightly shorter than the sum of covalent radii of two Ru atoms (2.8 Å)16 but is comparable to the reported Ru⋯Ru distances of 2.805(1) Å in [Ru3Cl12]4−,17a 2.775(1) Å in [LRuRuRuL]2+ (L = 1,4,7-tris(4-tert-butyl-2-mercaptobenzyl)-1,4,7-triazacyclononane),17b and 2.757(1) Å in [Ru3(aet)6]3+ (aet = 2-aminoethanethiol) complexes,17c for which RuIII⋯RuIII bonding interaction has been proposed.
![[RuIII3(TSA-H)2(TSA)4][NEt4] (1).](/image/article/2011/SC/c1sc00203a/c1sc00203a-f1.gif) |
| | Fig. 1
[RuIII3(TSA-H)2(TSA)4][NEt4] (1). | |
![Ball-and-stick representation of the X-ray crystal of 1 and its simplified core structure showing the linear S-bridged [Ru⋯Ru⋯Ru] moiety.](/image/article/2011/SC/c1sc00203a/c1sc00203a-f2.gif) |
| | Fig. 2 Ball-and-stick representation of the X-ray crystal of 1 and its simplified core structure showing the linear S-bridged [Ru⋯Ru⋯Ru] moiety. | |
Stability analysis
Complex 1 is fairly stable in PBS (pH 7.4)-DMSO (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution as revealed by less than 15% spectral change of its absorption peak at ∼480 nm for a time period of 72 h (Fig. S2, ESI†). In the presence of GSH, there are distinct changes in the UV-visible absorption spectrum of 1 in the PBS (pH 7.4)-DMSO (99:1, v/v) solution. The absorption peak with λmax of 480 nm is red shifted to 528 nm, which is concomitant with the formation a new absorption peak at ∼750 nm (Fig. S3, ESI†). This spectral change, revealed by incubation of a solution of 1 and GSH for 24 h followed by analysis using hybrid-time-of-flight (Q-TOF) tandem mass spectrometry, is attributed to the formation of adduct (or a loosely associated molecular species) between 1 and GSH (Fig. 3). Adducts of 1 with the oxidized glutathione (GSSG, Fig S4, ESI†) were also detected.
:1, v/v) solution as revealed by less than 15% spectral change of its absorption peak at ∼480 nm for a time period of 72 h (Fig. S2, ESI†). In the presence of GSH, there are distinct changes in the UV-visible absorption spectrum of 1 in the PBS (pH 7.4)-DMSO (99:1, v/v) solution. The absorption peak with λmax of 480 nm is red shifted to 528 nm, which is concomitant with the formation a new absorption peak at ∼750 nm (Fig. S3, ESI†). This spectral change, revealed by incubation of a solution of 1 and GSH for 24 h followed by analysis using hybrid-time-of-flight (Q-TOF) tandem mass spectrometry, is attributed to the formation of adduct (or a loosely associated molecular species) between 1 and GSH (Fig. 3). Adducts of 1 with the oxidized glutathione (GSSG, Fig S4, ESI†) were also detected.
![Q-TOF-MS (negative mode) of 1 (top) and 1 incubated with glutathione (GSH, middle) for 24 h. (Bottom) The simulated isotope patterns based on the [1-GSH]2− adduct.](/image/article/2011/SC/c1sc00203a/c1sc00203a-f3.gif) |
| | Fig. 3
Q-TOF-MS (negative mode) of 1 (top) and 1 incubated with glutathione (GSH, middle) for 24 h. (Bottom) The simulated isotope patterns based on the [1-GSH]2− adduct. | |
Cellular uptake
A major concern for sizable polynuclear complexes on therapeutic applications is their capability in passing through the cellular membrane of the target cells so as to reach the potential cellular target(s). Using cervical epithelioid carcinoma (HeLa) as an example, the intracellular ruthenium content of the cells treated with 1 has been determined. By means of inductively coupled plasma mass spectrometry (ICP-MS), 1.69 ng of ruthenium/cell (∼9800 ng of ruthenium in 5.8 × 106cells) was found in HeLa cells treated with 1 for 4 h. In a control experiment, significantly lower ruthenium content of 0.03 pg/cell was detected for HeLa cells treated with another trinuclear reference complex triruthenium dodecacarbonyl [Ru3(CO)12]. We reckon that the lipophilic moieties on the 2-thiosalicylic acid and/or thiosalicylate ligands together with the better aqueous solubility of 1 may play an important role in facilitating the cellular uptake.
In vitro cytotoxicity
As prompted by the significant cellular absorption, the cytotoxic activities of 1 towards a panel of human cancer cell lines including cervical epithelioid carcinoma (HeLa), breast cancer (MDA-MB-231), nasopharyngeal carcinoma (SUNE1) and hepatocellular carcinoma (HepG2), have been examined by means of a MTT assay.18 From the cytotoxicity profiles as depicted in Fig. 4, corresponding IC50 values of 1 (dose required for the inhibition of 50% cellular growth) towards the cancer cell lines are 7.3–18.2 μM, which are comparable to those of the clinically-used cisplatin [IC50 = 6.2 μM (HeLa), 13.6 μM (MDA-MB-231), 8.4 μM (SUNE1) and 17.2 μM (HepG2)]. For reference, the cytotoxicities of the TSA-H2 ligand and the precursor complex Ru(acac)3 were also examined. The TSA-H2 ligand is relatively non-toxic, with IC50 values of >100 μM towards the examined cell lines. The IC50 values of Ru(acac)3 were found to be ∼80 μM toward HeLa and SUNE1, and >100 μM towards the rest of the examined cell lines.
 |
| | Fig. 4 Cytotoxicity profiles of 1 towards various cell lines. | |
Using lung fibroblast cells (CCD-19Lu), the cytotoxicity of 1 toward non-cancerous originated cells was also assessed. The IC50 value was determined to be 32.1 ± 4.8 μM (Fig. 4), indicating that this complex is less cytotoxic towards the lung fibroblasts than that of the examined cancerous cells. Thus, 1 displays a 4.4-fold higher in vitro cytotoxicity to the human cervical epithelioid carcinoma HeLa cells than to the normal lung fibroblast cells.
Induction of apoptosis and cell-cycle arrest
We have examined the effects of 1 on cell-cycle progression in cancer cells by means of flow-cytometric analysis. Compared to the vehicle control, incubation of 1 at 3 and 10 μM in HeLa cells for 48 h did not induce apparent arrest of cells at either G0/G1, G2/M, S, or the sub-G1 apoptosis phase (Table 1). The result implies that 1 does not induce significant cell-cycle arrest including that in the S phase at the indicated dose concentrations of 48 h-treatment. In contrast, increasing the complex concentration of 1 to 30 μM significantly induced apoptotic cell death of the HeLa cells, as revealed from the increase of sub-G1 apoptotic phase from 1.2% (vehicle control) to 16.5% of the 1-treated HeLa cells. Since S-phase cell cycle is responsible for DNA replication and synthesis,19 the induced cytotoxicity of 1 may not directly account for the DNA damage. This is different from most of the reported anti-cancer ruthenium complexes which have been proposed to display their cytotoxic activities by DNA damage.5–7
Table 1
Cell-cycle analysis of 1 on HeLa cells
| |
Sub-G1
|
G0/G1 |
G2/M |
S |
| Percentage |
| Vehicle control |
1.2 ± 0.1 |
73.6 ± 6.3 |
9.1 ± 2.8 |
17.3 ± 3.1 |
|
1 at 0.3 μM |
1.2 ± 0.3 |
71.2 ± 6.1 |
11.4 ± 3.3 |
17.4 ± 2.7 |
|
1 at 10 μM |
1.1 ± 0.3 |
68.5 ± 5.9 |
13.4 ± 2.0 |
18.1 ± 3.4 |
|
1 at 30 μM |
16.5 ± 4.9 |
64.0 ± 7.7 |
13.4 ± 4.1 |
22.6 ± 5.9 |
UV-vis spectrophotometry showed that addition of calf-thymus DNA (ctDNA) to a solution of 1 does not cause apparent spectral change with [ctDNA]/[1] ratios up to 10 (Fig S5, ESI†). We also found that 1 does not directly induce strand break to DNA as revealed by the experiment of co-incubation of circular plasmid DNA with 1 for 24 h. The subsequent gel-electrophoretic separation of the DNA mixture showed that 1 neither induced plasmid DNA relaxation nor DNA strand break (Fig. S6, ESI†). Moreover, in vitro Comet20 (Fig. S7, ESI†) and BrdU-TUNEL21 (Fig. S8, ESI†) assays both revealed that 1 could not induce DNA strand breaks and damage in HeLa cells with incubation time up to 24 h at 30 μM. All of these results support that DNA is not the primary target of 1.
Attenuation of Wnt-β-catenin signaling pathway
An important component of Wnt-β-catenin signaling pathway is the effector protein β-catenin which could activate the expression of Wnt-targeted genes for cellular proliferation.13 In the absence of Wnt signaling, function of β-catenin is blocked by a destruction complex consisting of several proteins. In this study, oligonucleotide mircoarray analysis has provided clues for the anti-cancer mechanism of 1 on this signaling pathway, since two genes, namely Shisa2 and Tle4, related to the inhibition on the Wnt signaling were found to up-regulate differentially in response to 1 (Table 2). We have validated the changes in expression of these genes by means of reverse transcriptase-polymerase chain reaction (RT-PCR, Fig. S9, ESI†). The RT-PCR experiment further confirmed that treatment of 1 on HeLa cells attenuates the Wnt-β-catenin signaling by suppression of the gene expression on Wnt5α and β-catenin, and concomitantly, on cyclin D1 (Fig. 5, left). By Western blot experiments, the protein expression levels of Wnt5α and β-catenin have also been examined. As shown in Fig. 5 (right), both the protein expression levels of Wnt 5α and β-catenin decreased with time. Taking these results altogether, complex 1 attenuates the Wnt-β-catenin signaling pathway in both transcriptomics and proteomics levels.
Table 2 Selected genes differentially expressed in response to 1 as determined by DNA microarray in HeLa cells
|
Probe set ID |
Entrez gene |
Gene title |
Gene symbol |
Fold change |
| 206375_s_at |
8988 |
Heat shock 27kDa protein 3 |
Hspb3
|
+1.79 |
| 230493_at |
387914 |
Shisa homolog 2 |
Shisa2
|
+1.69 |
| 235765_at |
7091 |
Transducin-like enhancer of split 4 |
Tle4
|
+1.57 |
| 202191_s_at |
8522 |
Growth arrest-specific 7 |
Gas7
|
+1.82 |
| 1558214_s_at |
1495 |
Catenin (cadherin-associated protein), alpha 1, 102kDa |
Ctnna1
|
+1.71 |
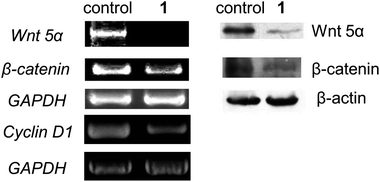 |
| | Fig. 5 (Left) Expression profiles of Wnt 5α, β-catenin and Cyclin D1 in HeLa cells in response to 1 at 30 μM for 24 h by RT-PCR experiment. (Right) Western blotting experiment showing the protein expression levels of Wnt 5α and β-catenin of HeLa cells treated with 1 at 30 μM for 24 h. | |
Although the Wnt signaling pathway has been identified for over two decades, in this work, we were attracted to some recent intriguing findings including the correlation of this pathway with reactive oxygen species (ROS) and with the anti-metastatic activities in cancer cells.
ROS formation
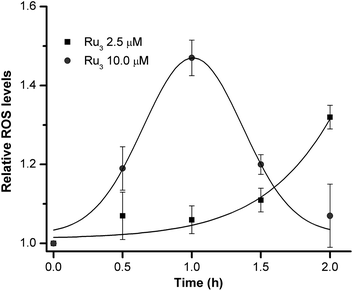
Miki and co-workers in a recent paper have demonstrated that some reactive oxygen species (ROS) may help modulate the Wnt-β-catenin signaling pathway.22 In the present study, treatment of 1 on HeLa cells was found to time- and dose-dependently induce ROS formation. The intracellular production of ROS upon treatment of HeLa cells with 1 was evaluated using the 5-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate (CM-H2DCFDA) approach.23 As depicted in Fig. 6, the relative ROS levels compared to the untreated control as determined from the cell-associated fluorescence reveal that 1 could moderately induce ROS formation (∼45% enhancement) at concentration of 10 μM. The fluorescence signal was found to increase sharply from 0 to 1 h of incubation, and was in plateau upon treatment of 1 for 1 h. Prolonged incubation of 1 at this concentration led to significant reduction in fluorescent intensity. Lowering the concentration of 1 to 2.5 μM initially had no significant enhancement in ROS formation until 1.5 h of incubation had reached.
We found that the induction of ROS by 1 is crucial for the attenuation of the Wnt-β-catenin signaling pathway. As shown in the Western Blot experiments, pretreatment of the HeLa cells by an ROS scavenger N-acetyl-L-cysteine (NAC) significantly altered the attenuation effect by 1 on the protein expressions of Wnt5α and β-catenin (Fig. 7). Yet, the possibility on the direct induction of apoptosis triggered by the ROS cannot be ruled out. Our preliminary study also revealed that co-treatment of an ROS inducer H2O2 with 1 led to an apparent synergistic effect on the cytotoxicity toward HeLa cells (Fig. S10, ESI†).
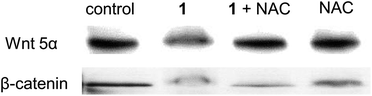 |
| | Fig. 7 Western blotting experiment showing the protein expression levels of Wnt 5α and β-catenin of HeLa cells treated with 1 at 30 μM in the presence or absence of ROS scavenger N-acetyl-L-cysteine (NAC) for 24 h. | |
Anti-migration activity
The results from the RT-PCR and Western blotting experiments as shown in Fig. 5 reveal that 1 could inhibit the gene and/or protein expressions on Wnt 5α. Together with the up-regulated genes of Gas7 and Ctnna1 (Table 2 and Fig. S9, ESI†), all these components have recently been proven to associate with cellular proliferation and migration in endothelial and various cancer cells.13 To determine whether 1 would inhibit migration of the cancer cells, wound-healing experiment was performed. We found that 1 at its sub-toxic concentrations of 0.3 μM and 1 μM significantly inhibited cell migration (Fig. S11, ESI†). At these concentrations, 1 markedly inhibited migration of the HeLa cells with an incubation time of 8 h compared with the vehicle control. The inhibitory effect was more prominent with prolonged treatment for 24 h. Thus, complex 1 at its sub-toxic concentration could effectively inhibit cellular migration of HeLa cells, suggestive of its potential anti-metastatic activity.
Conclusion
An anti-cancer active trinuclear ruthenium(III) complex with the non-toxic 2-thiosalicylic acid ligands has been prepared and structurally characterized. This complex forms adduct(s) with the biological reductant glutathione (GSH), induces apoptosis, and reduces both transcriptomic and protein expression levels of central components regarding the Wnt-β-catenin signaling pathway. This work highlights the potential application of polynuclear metal complexes in the development of new anti-cancer agents targeting the Wnt-β-catenin signaling pathway.
Acknowledgements
We acknowledge support from the ITF-Tier 2 project (ITS/134/09FP) administrated by Innovation and Technology Commission (HKSAR, China), and the Areas of Excellence Program (AoE/P-10/01) administrated by University Grants Council (HKSAR, China).
Notes and references
-
(a) S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 14689 CrossRef CAS;
(b) C.-M. Che and F.-M. Siu, Curr. Opin. Chem. Biol., 2010, 14, 255 CrossRef CAS;
(c) S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089 CrossRef CAS;
(d) P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197 CrossRef CAS;
(e) R. W.-Y. Sun, D.-L. Ma, E. L.-M. Wong and C.-M. Che, Dalton Trans., 2007, 4884 RSC;
(f) S. J. Lippard, Nat. Chem. Biol., 2006, 2, 504 CrossRef CAS; H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3563 CrossRef CAS;
(g) C. Orvig and M. J. Abrams, Chem. Rev., 1999, 99, 2201 CrossRef CAS.
-
(a) G. E. Atilla-Gokcumen, L. Di Costanzo and E. Meggers, JBIC, J. Biol. Inorg. Chem., 2011, 16, 45 CrossRef CAS;
(b) S. P. Mulcahy, K. Gründler, C. Frias, L. Wagner, A. Prokop and E. Meggers, Dalton Trans., 2010, 39, 8177 RSC;
(c) E. Meggers, Chem. Commun., 2009, 1001 RSC.
- Selected examples:
(a) S. J. Berners-Price, Angew. Chem., Int. Ed., 2011, 50, 804 CrossRef CAS;
(b) R. W.-Y. Sun, A. L.-F. Chow, X.-H. Li, J. J. Yan, S. S.-Y. Chui and C.-M. Che, Chem. Sci., 2011, 2, 728 RSC;
(c) J. Liu, C.-H. Leung, A. L.-F. Chow, R. W.-Y. Sun, S.-C. Yan and C.-M. Che, Chem. Commun., 2011, 47, 719 RSC;
(d) L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS;
(e) J. H. Schiller, D. Harrington, C. P. Belani, C. Langer, A. Sandler, J. Krook, J. Zhu and D. H. Johnson, N. Engl. J. Med., 2002, 346, 92 CrossRef CAS.
- Selected examples:
(a) K. H.-M. Chow, R. W.-Y. Sun, J. B. B. Lam, C. K.-L. Li, A. Xu, D.-L. Ma, R. Abagyan, Y. Wang and C.-M. Che, Cancer Res., 2010, 70, 329 CrossRef CAS;
(b) V. Milacic and Q. P. Dou, Coord. Chem. Rev., 2009, 253, 1649 CrossRef CAS;
(c) R. W.-Y. Sun and C.-M. Che, Coord. Chem. Rev., 2009, 253, 1682 CrossRef CAS;
(d) A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692 CrossRef CAS;
(e) D. Fregona, L. Ronconi and D. Aldinucci, Drug Discovery Today, 2009, 14, 1075 CrossRef;
(f) C.-M. Che, R. W.-Y. Sun, W.-Y. Yu, C.-B. Ko, N. Zhu and H. Sun, Chem. Commun., 2003, 1718 RSC;
(g) C. F. III Shaw, Chem. Rev., 1999, 99, 2589 CrossRef CAS.
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511 CrossRef CAS.
-
(a) I. Kostova, Curr. Med. Chem., 2006, 13, 1085 CrossRef CAS;
(b) E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525 CrossRef CAS.
-
(a) S. J. Dougan, A. Habtemariam, S. E. McHale, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11628 CrossRef CAS;
(b) L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2007, 251, 1633 CrossRef CAS.
- R. W.-Y. Sun, M. F.-Y. Ng, E. L.-M. Wong, J. Zhang, S. S.-Y. Chui, L. Shek, T.-C. Lau and C.-M. Che, Dalton Trans., 2009, 10712 RSC.
- F. A. Cotton, C. Lin and C. A. Murillo, Acc. Chem. Res., 2001, 34, 759 CrossRef CAS.
- E. L.-M. Wong, R. W.-Y. Sun, N. P.-Y. Chung, C.-L. S. Lin, N. Zhu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 4938 CrossRef CAS.
- P. N. Rao, M. L. Smith, S. Pathak, R. A. Howard and J. L. Bear, J. Natl. Cancer Inst., 1980, 64, 905 CAS.
- J. D. Roberts, J. Peroutka and N. Farrell, J. Inorg. Biochem., 1999, 77, 51 CrossRef CAS.
-
(a) H. Clevers, Cell, 2006, 127, 469 CrossRef CAS;
(b) T. Reya and H. Clevers, Nature, 2005, 434, 843 CrossRef CAS.
- S. Dihlmann, A. Siermann and M.v. K. Doeberitz, Oncogene, 2001, 20, 645 CrossRef CAS.
- B. A. Teicher, S. A. Holden, T. S. Herman, E. A. Sotomayer, V. Khandekar, K. W. Rosbe, T. W. Brann, T. T. Korbut and E. III Frei, Int. J. Cancer, 1991, 47, 252 CAS.
- Tables of covalent and van der Waals radii are available at URL: http://www.ccdc.cam.ac.uk/products/csd/radii.
-
(a) A. Bino and F. A. Cotton, J. Am. Chem. Soc., 1980, 102, 608 CrossRef CAS;
(b) B. Albela, E. Bothe, O. Brosch, K. Mochizuki, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 1999, 38, 5131 CrossRef CAS;
(c) N. Matsuura, A. Igashira-Kamiyama, T. Kawamoto and T. Konno, Inorg. Chem., 2006, 45, 401 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS.
- E. A. Harrington, J. L. Bruce, E. Harlow and N. Dyson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11945 CrossRef CAS.
- D. W. Fairbairn, P. L. Olive and K. L. O'Neill, Mutat. Res., Rev.Genet. Toxicol., 1995, 339, 37 Search PubMed.
- M. A. Kern, D. Schubert, D. Sahi, M. M. Schöneweiß, I. Moll, A. M. Haugg, H. P. Dienes, K. Breuhahn and P. Schirmacher, Hepatology, 2002, 36, 885 CAS.
- Y. Funato, T. Michiue, M. Asashima and H. Miki, Nat. Cell Biol., 2006, 8, 501 CrossRef CAS.
- F. Turturro, E. Friday and T. Welbourne, BMC Cancer, 2007, 7, 96 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedure and characterization of the trinuclear ruthenium complex; experimental procedures for X-ray crystallography, cellular uptake, cell culture, cytotoxicity evaluation, flow cytometric analysis, DNA assays, RT-PCR assay, Western blotting, ROS assay and wound-healing assay. Fig. S1: Magnetic property of 1; Fig. S2: Stability of 1 in solution; Fig. S3: Stability of 1 in PBS in the presence of GSH; Fig. S4: ESI spectrum of the 1-GSSG adduct; Fig. S5: Absorption titration experiment of 1 with ctDNA; Fig. S6: gel-electrophoretic separation of the DNA mixtures; Fig. S7: Comet Assay; Fig. S8: BrdU-TUNEL assay; Fig. S9: RT-PCR experiment; Fig. S10: Cytotoxicity profile of 1 with increasing concentration of H2O2; and Fig. S11: wound-healing assay. CCDC reference numbers 804342. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00203a |
| ‡ These authors contribute equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.