Two-dimensional supramolecular chemistry on surfaces
Anna G.
Slater (née Phillips)
a,
Peter H.
Beton
b and
Neil R.
Champness
*a
aSchool of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Neil.Champness@nottingham.ac.uk; Fax: (+44)115-9513563; Tel: (+44)115-9513505
bSchool of Physics and Astronomy, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 13th June 2011
Abstract
Self-assembly of two-dimensional supramolecular arrays on surfaces represents a significant challenge to chemists, materials scientists and physicists. This article highlights advances in using supramolecular interactions, particularly hydrogen bonding, to self-assemble such two-dimensional arrays on surfaces. Scanning-probe microscopies, particularly scanning tunnelling microscopy (STM), can be used to determine the precise molecular arrangement of the self-assembled structures allowing insight into the self-assembly process at the molecular level. The use of such supramolecular assemblies to trap guest species, mimicking host–guest chemistry in the solution phase, will also be discussed. Such images provide great insight into the advantages and restrictions of working in two dimensions in comparison to the solution phase or the solid state.
Introduction
The use of self-assembly processes to create nanoscale architectures lies at the heart of supramolecular chemistry.1 The vast majority of supramolecular chemistry research has taken place in the solution phase, creating a plethora of intricate structures with an increasing number of potential applications.2 Solid-state supramolecular chemistry is also a highly developed research field and is intimately linked with the wider field of crystal engineering.3 However, a particularly intriguing and relatively new field of research is that of surface-based supramolecular chemistry that inherently favours the formation of two-dimensional structures.4–9 Surface-based chemistry has long been utilised for assembling arrays of molecules. Perhaps the most widely studied area of such chemistry is the development of self-assembled monolayers (SAMs) of thiolate molecules adsorbed onto Au(111) substrates.10 The development of Au-thiolate systems, and related approaches to molecular tethering,11 has been extensive and lies at the heart of many studies that attach molecules and in some cases supramolecular arrays to surfaces.12Deposition of molecules onto surfaces often leads to close-packed arrays that have well-defined arrangements, however, such arrays typically rely on simple van der Waals interactions and simple geometric preferences rather than stronger, well-defined, supramolecular interactions. This article will focus on studies where molecules are specifically designed in an attempt to control relative molecular organisation, a concept that lies at the heart of supramolecular chemistry and the molecular nanosciences. In particular, the article will focus on the use of hydrogen-bonding to form extended arrays that propagate in parallel to the plane of a surface substrate. In addition to, and sometimes in combination with, hydrogen bonding other interactions have been used to prepare surface-based supramolecular assemblies including metal–ligand coordination interactions,12,13 dipolar coupling,14halogen bonding15 and van der Waals interactions.7–9,16 Two-dimensional self-assembly of molecules on surfaces, when combined with scanning probe imaging techniques, provides direct evidence of the potential of this approach. The exploitation of porous two-dimensional architectures to trap diffusing species as guests, in a similar fashion to porous architectures constructed in three-dimensional solids,17 is also highlighted.
Surface-based supramolecular chemistry differs from more traditional supramolecular chemistry in a number of ways. Most importantly, the surface defines a two-dimensional boundary upon which the self-assembly process takes place. However, the surface cannot be considered as passive and can interfere with the self-assembly process. Typically the surface forms weak interactions with the molecules in preferred adsorption sites thus influencing the self-assembly process. It is also worth noting that surfaces are far from perfect structures, and surface step-edges and defects are often found to interfere with the target supramolecular structure, in some instances acting as a starting point for self-assembly. A range of surfaces has been used for such studies but perhaps the most commonly studied systems include Au(111),18 and highly-oriented pyrolytic graphite (HOPG),19 due to their stability and relative ease of use. More exotic surfaces are open to the studies and recent investigations using graphene16 are likely to spur further studies on this highly topical material.
Two-dimensional hydrogen-bonded networks on surfaces
Supramolecular chemistry has developed a large toolkit of interactions that can be exploited to synthesise self-assembled structures. Hydrogen bonds have always been a focus of attention in supramolecular chemistry with much inspiration being drawn from nature.Amongst the most commonly studied synthons used in supramolecular chemistry are those that exploit carboxylic acids. Carboxylic acids are very versatile and form robust hydrogen bonding interactions20 with other carboxylic acids, as well as a range of hydrogen bond acceptor groups. Carboxylic acids have been used in surface supramolecular chemistry from the earliest stages of the field21–29 and remain one of the most widely studied building-blocks. Although one-dimensional chains are readily formed using simple ditopic carboxylic acid-containing molecules,22 this article will focus on two-dimensional structures where building-blocks capable of more than two interactions are required to form a larger array.
The simplest approach to creating a surface-based self-assembled structure is to use a single molecule capable of creating an extended supramolecular network without the need to add any additional agents. An early example of the formation of a unimolecular supramolecular structure mediated by hydrogen bonding was reported by Griessl et al.23 using 1,3,5-benzenetricarboxylic acid. The supramolecular structure is formed by the deposition of the molecule onto a HOPG substrate in UHV conditions. Imaging using STM reveals precise details of the molecular arrangement that is adopted as a result of the self-assembly process (Fig. 1). It can be clearly seen that the observed configuration is formed in preference to an alternative close-packed arrangement confirming the importance of intermolecular hydrogen-bonding between carboxylic acids in producing the structure. The system adopts the well-known R22(8) CO2H⋯HO2C interaction, familiar to many supramolecular chemists, leading to the formation of the expected porous ‘chicken-wire’, or honeycomb, network structure (Fig. 1). However the influence of the underlying substrate is well-demonstrated by this system as, in addition to the honeycomb structure, a second, more dense, ‘flower’ arrangement is also observed by STM measurements (Fig. 1). This alternative self-assembled structure results from the formation of R33(12) supramolecular synthons formed by three carboxylic acid moieties from distinct 1,3,5-benzenetricarboxylic acid molecules. It is envisaged that a contributing factor in the formation of the ‘flower’ structure is the higher molecular density on the surface, maximising energy gained through adsorbate-substrate interactions. Interestingly, images of these structures could not be obtained at room temperature, potentially due to weak interactions between the molecules and the substrate and resulting high molecular mobility.
 | ||
| Fig. 1 Open network arrangements of 1,3,5-benzenetricarboxylic acid on graphite under UHV at low temperature. a), c) the ‘chicken-wire’ structure. b), d) the ‘flower’ arrangement.23 Reproduced with permission from Wiley-VCH Verlag GmbH and Co. KGaA. | ||
A subsequent study has investigated an extended analogue of 1,3,5-benzenetricarboxylic acid, 1,3,5-tris(4-carboxyphenyl)benzene, on HOPG, but deposited from a range of alkyloic acids.24 The study also reveals the formation of a honeycomb lattice at low temperatures but a more densely packed phase at higher temperatures.24 The phase transition and the transition temperature were found to depend on the nature of the solvent and molecular concentration. The authors suggest that the co-adsorption of solvent molecules within the honeycomb structure stabilizes the nominally porous structure at low temperatures, but, upon elevation of the temperature the weakly bound solvent molecules desorb initiating the transition to the more densely packed and thermodynamically favoured phase.
The range of potential carboxylic acid building-blocks is extensive but one class of tetracarboxylic acids that we have studied25,29 have provided highly unusual examples of self-assembled structures, extending research pathways in the field. Initial studies by Wuest et al. investigated the adsorption of tetracarboxylic acid molecules on an HOPG surface revealing the formation of a range of structural motifs.25 Interestingly Wuest observed that two polymorphic arrangements are possible for such systems, a “parallel” motif and a Kagomé network (Fig. 2).
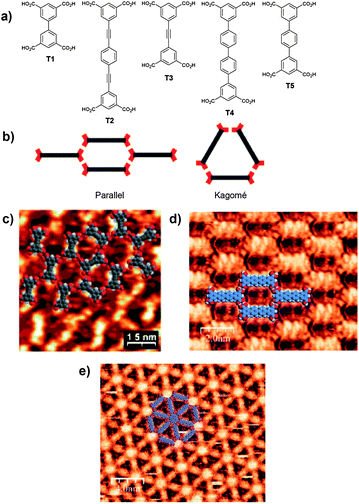 | ||
| Fig. 2 a) Structures of tetracarboxylic acid tectons T1–T5; b) Representation of the ‘parallel’ and ‘Kagomé’ network observed for self-assembled tetracarboxylic acid tectons, red crescents represent isophthalic acid moieties. STM images of self-assembled arrays of tetracarboxylic acid tectons adsorbed on HOPG; c) T3 forming adopting both the ‘parallel’ and ‘Kagomé’ arrangements;25 d) T4 forming the ‘parallel’ arrangement and following addition of the guest molecule coronene; e) The ‘Kagomé’ network.26 b), d) and e) Reproduced with permission from the Royal Chemical Society. c) Reproduced with permission from the American Chemical Society. | ||
Whereas the shortest tetracarboxylic acid tecton, biphenyl-3,3′′′,5,5′′′-tetracarboxylic acid T1, forms the parallel network the significantly longer tecton, 1,4-diethynlphenylenedi-3′,3′′′,5′,5′′′-tetracarboxylic acid T2, produces the Kagomé network. The third tecton studied, diphenylethyne-3,3′,5,5′-tetracarboxylic acid T3, presented what was termed a ‘frustrated’ system in which localised regions of both parallel and Kagomé like arrangements were observed (Fig. 2), with smooth transitions from one arrangement to the other.
In our studies we have observed that quaterphenyl-3,3′′′,5,5′′′-tetracarboxylic acid T4, adopts the R22(8) intermolecular CO2H⋯HO2C interaction in a similar manner to the other tetracarboxylic acid tectons forming a porous two-dimensional structure (Fig. 2).26 In this case the nature of the observed structure can be modified by addition of a guest template molecule, in this instance coronene, which fits within a cycle formed by six isophthalic acid moieties. Addition of coronene to the solution either prior or subsequent to deposition of the tetracarboxylic acid species results in the adoption of a more open porous structure with coronene sitting in a hexa-isophthalic acid pore as predicted (Fig. 2). The process of guest-induced transformation of the resulting supramolecular structure mirrors guest-induced fit processes typically observed in solution-phase supramolecular chemistry.30 In related studies, other tetracarboxylic acid building-blocks have been used to act as hosts for guest molecules, including a particularly interesting example that entraps fullerene and endohedral fullerene species in regular arrays.29
Interestingly, studies of terphenyl-3,3′′,5,5′′-tetracarboxylic acid T5, adsorbed onto HOPG similarly leads to the formation of a two-dimensional hydrogen-bonded structure that utilizes R22(8) intermolecular hydrogen-bonding interactions. However, in this instance rather than forming an ordered structure, in the paradigm familiar to supramolecular chemists, the relative position of molecules within the array is random and reminiscent of dynamically-arrested systems such as glasses (Fig. 3).27 The intriguing structure arises from the similar stabilities of five distinct hexagonal junctions which are formed from three, four, five, or six molecules. The different arrangements arise as a result of the dimensions of the molecule whose length (d2, 8.7 Å) is similar to the distance across the R22(8) intermolecular hydrogen-bonding interaction (d1, 9.6 Å), facilitating the formation of the range of observed configurations. The structure can also be interpreted as the first physical example of a random, entropically stabilized, rhombus tiling.
 | ||
| Fig. 3 a) STM image of a typical area of terphenyl-3,3′′,5,5′′-tetracarboxylic acid (T5) network at the nonanoic acid/HOPG interface. The groups of three phenyl rings of the molecule backbone appear as bright features in the image. The hexagonal orientational order of the structure is indicated by the group of blue dots in the lower right-hand corner of the image, marking the location of pores in the network. b) Diagrams representing the five possible arrangements of T5 molecules around a network pore, accompanied by magnified STM image examples of each pore type. The locations of the magnified regions are marked in a) by blue dashed squares.27 c) The calculated distances between two phenyl rings of different TPTC molecules measured across a carboxylic acid…carboxylic acid hydrogen bond (d1), and the distance between the two end phenyl rings of a single T5 molecule (d2). Reproduced with permission from the AAAS. | ||
The random structure formed by T5 on HOPG is also able to trap guest molecules.28C60 can be adsorbed into the random array with preferential adsorption into one of the five possible pores. The images reveal a strong preference for adsorption in pores of type A (Fig. 4), which make up approximately 40% of pores in the initial layer but ca. 76% of pores that trap a C60 molecule. Pores B–E show a lower-than-expected C60 trapping occupancy confirming the preferential adsorption of C60 in pore A. Calculations demonstrate that adsorption in pore A is favoured because of the higher proportion of phenyl edges in this configuration. Moreover STM images reveal that in regions of the network where C60 molecules have been adsorbed a second supramolecular network is assembled sitting over the initial layer, thus forming a bilayer structure (Fig. 4). This was the first time that growth of a bilayer has been observed in such supramolecular assemblies. The effect is clearly a result of templating by the spherical C60 molecule; indeed, the formation of the second layer and C60 entrapment are co-dependent. Furthermore, the addition of coronene to the sample results in displacement of the entrapped C60 by the flat coronene molecule and concomitant removal of the second supramolecular layer.
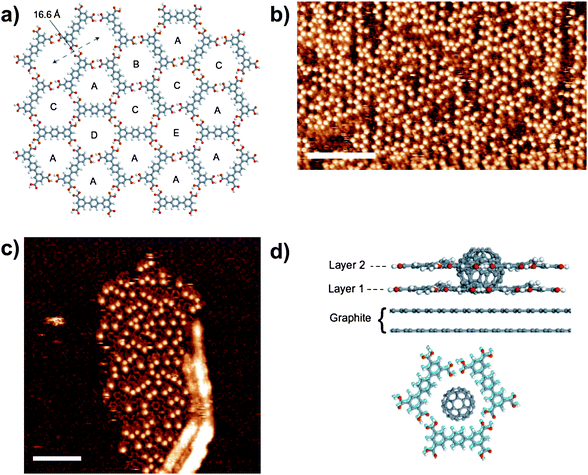 | ||
| Fig. 4 a) Molecular schematic of a section of T5 network that highlights the hexagonally ordered network of pores, A–E; b) STM image of an area of T5 network ca. 24 h after deposition of C60. The locations of C60 are clearly visible as the bright spots in the image; the underlying T5 network structure is not visible. Scale bar = 160 Å; c) STM image of T5 network immediately after C60 deposition. An island of C60 and bilayer T5 network grows away from a surface defect. The initial layer of T5 network is visible with an altered contrast and the T5 molecules in the second layer appear with the long axis of the molecules as bright, rod-like features. Scale bar = 110 Å. d) Side-view of the C60–bilayer network that consists of two overlying pores of type A and a view perpendicular to the surface plane with the C60 placed at its minimum-energy position for both the first- (light blue) and second-layer pores (grey). The two layers are displaced slightly with respect to each other, which aids clarity, but in addition is expected on the basis of calculations. Reproduced with permission from the Nature Publishing Group.28 | ||
Supramolecular chemistry using hydrogen bonding interactions is very well developed and therefore a number of asymmetric supramolecular synthons can serve as inspiration for surface supramolecular chemistry. The triple hydrogen bonding interaction between 2,6-di(acetylamino)pyridines and imide groups has been identified as one of the most reliable supramolecular synthon31 and has been widely exploited in supramolecular chemistry in the formation of tapes,32–34 rosettes35 and capsules36 in the solution phase,32,35,36 the solid-state,33,34 and at interfaces.37
Thus, we have developed a family of supramolecular structures built using interactions between perylene-tetracarboxylic diimide (PTCDI) species and melamine. The prototypical structure formed between PTCDI and melamine38 was initially prepared by co-deposition of the two species onto Ag-Si(111)√3x√3R30° [named Ag/Si(111) from here on] under UHV conditions. The resulting structure is a honeycomb network formed via triple hydrogen bonds adopted between the imide moieties of the PTCDI molecules and each face of melamine (Fig. 5). Network formation was achieved by the sequential deposition of PTCDI and melamine, followed by an annealing step at ∼100 °C. Annealing provides sufficient thermal energy for molecules to detach from PTCDI islands and diffuse across the surface, resulting in the desired network structure. The network was found to be commensurate with the underlying Ag/Si(111) surface, indicating an influence from the substrate in the formation of the network.
 | ||
| Fig. 5 a) A schematic of the PTCDI-melamine junction, showing the 9 hydrogen bonds that make up the structural node as dashed red lines; b) STM image of the PTCDI-melamine network on Ag/Si(111); inset, high resolution view of the Ag/Si(111) surface; c) Schematic of the network showing the registry with the hexagonal substrate; d) STM image of fullerenes trapped within the pores of the hexagonal network, seen as bright white features; e) A schematic diagram of a C60 heptamer sitting within a pore.38 Reproduced with permission from the Nature Publishing Group. | ||
The pores formed by the network, with a cross section of 2.4 nm, are capable of housing several large molecules. Indeed the network has been shown to host molecular guests, notably C6038–40 (Fig. 5) but also C84,41 and more recently Mn12O12(O2CCH3)16(H2O)4 clusters.42 The deposition of C60 by sublimation onto the hexagonal network leads to the formation of heptameric C60 clusters within the pores of the network, with a compact hexagonal arrangement of fullerenes aligned parallel to the principle axes of the Ag/Si(111) surface (Fig. 5). The arrangement of the C60 clusters was found to differ from close-packed fullerene, which does not align with the principle axes of the underlying Ag/Si(111) surface or form heptamers, demonstrating the templating and stabilizing effects of the network.38
The PTCDI-melamine network discussed above32 can similarly be prepared on a Au(111) surface43 leading to a honeycomb arrangement analogous to that observed on Ag/Si(111). However, a parallelogram phase was also observed on this surface after annealing at higher temperatures, which has the same stoichiometric ratio as the honeycomb structure but is more compact.39 The parallelogram network also acts as a trap for C60 molecules39,40 but, due to the restricted cavity size, C60 dimers are observed. Similar studies have demonstrated entrapment of Lu@C82 by the PTCDI/melamine parallelogram network.44 Interestingly the same parallelogram network also traps two decanethiol molecules which sit parallel to the surface under UHV conditions.45 The entrapment of a significantly different and potentially more functional molecule demonstrates that such networks have the potential for trapping a range of different species. However it is interesting to note that adsorption of decanethiol onto the PTCDI-melamine arrays lead to the destruction of the honeycomb phase but not the parallelogram phase, suggesting that the latter is preferentially stabilised by the thiol guest.45
The ability of PTCDI-melamine networks to adsorb guest molecules can also be modified by molecular design through functionalisation of the PTCDI with groups in what is known as the ‘bay’ area of such molecules (Fig. 6).46,47 Thus by simple crowding of the pores through chemical functionalisation, guest entrapment can be controlled such that networks trap individual C60 molecules within regularly spaced pores.
 | ||
Fig. 6 a) STM image and b) model of a self-assembled network of 1,7-bis(4-benzoic acid)-3,4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9,10-perylenetetracarboxylic diimide encapsulating regularly spaced individual C60 molecules;47 c) PTCDI molecules functionalised in the ‘bay region’ used to prepare self-assembled arrays with restricted pore dimensions.46,47 Reproduced with permission from the Royal Chemical Society. :9,10-perylenetetracarboxylic diimide encapsulating regularly spaced individual C60 molecules;47 c) PTCDI molecules functionalised in the ‘bay region’ used to prepare self-assembled arrays with restricted pore dimensions.46,47 Reproduced with permission from the Royal Chemical Society. | ||
The studies outlined in the preceding paragraphs are all performed in UHV conditions but a report by Buck et al. demonstrates that the PTCDI-melamine network can be assembled on a Au(111) surface from solution and used for a variety of further applications, particularly as a versatile patterning tool.48,49 Interestingly the pores of the network can be used to host a range of different thiol molecules; adamantanethiol, ω-(4′-methylbiphenyl-4-yl)propane thiol and dodecanethiol have all be studied, and the studies demonstrate that the adsorbed thiol species sit perpendicular to the surface, in contrast to related studies in UHV conditions.45 The islands of adsorbed thiols, which may be considered as confined SAMs, open up a variety of possibilities as the study demonstrates a link between the self-assembled hydrogen-bonded structures discussed throughout this article and the well-established field of thiol SAMs.10 Buck et al. demonstrated the use of the confined adamantanethiol SAMs to guide Cu atom deposition within the PTCDI/melamine network pores through underpotential deposition (UPD), such that only areas of the surface covered by thiols were successfully covered with Cu.48 A further study demonstrated that the PTCDI-melamine network acts as a diffusion barrier for Cu adatoms.50 The use of the hydrogen-bonded array to direct the formation of islands of Cu atoms demonstrates that the network is stable to further fabrication steps and indicates a wider applicability of such an approach to the preparation of nanostructures on small length scales, typically 2–5 nm.
The substrate clearly influences the self-assembly process and thus many possible strategies to influence the final structure can be envisaged. A notable class of substrates are those that exhibit moiré patterns, introducing distinct adsorption sites on the surface and therefore offering the possibility of influencing self-assembly. Such surfaces include graphene and BN ‘nanomesh’ monolayers grown on Rh(111) crystals.51,52 The assembly of PTCDI and related derivatives, 1,7-dipropylthio-perylene-3,4:9,10-tetracarboxydiimide (DP-PTCDI)47 and 1,7-di(butyl)-coronene-3,4:9,10-tetracarboxylic acid diimide (DB-CTCDI),53 have been studied on graphene and the structure of the resulting structures determined using STM.54 Two distinct types of array are observed involving distinct junctions formed by either two or three diimide molecules (Fig. 7). Thus, PTCDI forms rows up to 25 nm in length, adopting only simple dimeric hydrogen-bonding arrangements that run parallel to the principal directions of the surface. The molecular arrangement on the graphene superstructure differs significantly from that observed for a graphite substrate, on which close-packed three-dimensional islands are formed.55 The importance of commensurability between the molecular dimensions and the moiré periodicity is demonstrated by comparison with PTCDI adsorption on a BN “nanomesh” monolayer. The BN monolayer is isoelectronic with graphene, and also displays a moiré pattern but with a slightly larger periodicity (3.2 nm) which is not commensurate with the molecular dimensions of PTCDI. Thus, on BN individual, isolated, PTCDI molecules are trapped in energy minima associated with the moiré pattern demonstrating the influence of the underlying substrate on supramolecular assembly. Functionalisation of the PTCDI core in DP-PTCDI also leads to commensurate rows, but, compared to PTCDI, there are fewer examples of pairs of parallel rows and there are many more threefold hydrogen-bonded junctions following self-assembly on graphene. The ratio of dimer:trimer junctions is 75:25 for DP-PTCDI in contrast to less than 1% of junctions being trimers for PTCDI. Further extension of the PTCDI core in the coronene-3,4:9,10-tetracarboxylic acid diimide species DB-CTCDI leads to domination of the self-assembly process by threefold junctions, with no linear dimers being identified unambiguously. The array of trimers results in a honeycomb arrangement of molecules which is aligned with the graphene monolayer superstructure and encloses the areas of bright contrast arising from the moiré pattern.
 | ||
| Fig. 7 Dimer and trimer hydrogen-bonded junctions observed for PTCDI derivatives, in this case DP-PTCDI is represented. | ||
It is also possible to modify the structure of self-assembled systems by simple molecular design principles. Thus a range of molecules can be envisaged that exploit the same supramolecular synthons used in the PTCDI-melamine system. For example the interaction formed between cyanuric acid and melamine, and derivatives of the two compounds, exploits the same imide-diaminopyridine supramolecular synthon as PTCDI-melamine and represents one of the most extensively studied structural motifs in supramolecular chemistry.56 Thus the cyanuric acid-melamine network has been studied on both the Ag/Si(111)57 and Au(111)58 surfaces and the anticipated 1:1 cyanuric acid-melamine array is observed. Interestingly, the cyanuric acid-melamine complex can be formed directly, without traces of the single component cyanuric acid islands, by simultaneous deposition of cyanuric acid and melamine, indicating a significant preference for the cyanuric acid-melamine network under the conditions used.57
However, studies involving modification of component building blocks are often not always as simple as may be imagined due to the influential role of the underlying surface. As discussed above surface registry can affect network formation. Thus attempts to prepare an array analogous to the PTCDI-melamine network using 1,4,5,8-naphthalenetetracarboxylic diimide (NTCDI), the single component assembly of which has been studied,59 does not lead to an analogous honeycomb structure. Indeed, the lack of commensurability between the hypothetical NTCDI-melamine honeycomb array and the Ag-Si(111) surface results in destabilisation of such a network and leads to the formation of a range of other motifs. These alternative motifs do not form long range networks indicating that the self-assembly process does not have a strong preference for a single product array.60
When performing experiments at the solution/solid interface the self-assembly process is further complicated in comparison to UHV experiments. The presence of solvent introduces the possibility of molecular desorption from the surface, introducing an equilibrium between adsorbed and solvated molecules. Additionally, surface adsorption of solvent molecules provides a competitive process for adsorption and self-assembly of target species. Samori et al. have elegantly demonstrated the issues that can arise in such situations with multi-component systems.61,62 The self-assembly of a range of diimide molecules and melamine on an HOPG surface were studied following deposition from a 1,2,4-trichlorobenzene/dimethylsulfoxide solution. The formation of hexagonal networks would be anticipated for combinations of such systems, and are observed, but detailed studies of concentration variation leads to the adoption of a range of structures. The nature of the structures formed by combinations of diimide molecules with melamine ranges from open hexagonal networks to close-packed structures and importantly, varies with concentration of both components, leading to a phase diagram of polymorphs for the systems. The interaction between 2,6-di(acetylamino)pyridines and imide groups has also been exploited to assemble a range of different chemical functionalities in discrete surface-based supramolecular entities.51
The potential choices of hydrogen-bonding synthons are extensive and a range of systems have been explored.63DNA-based interactions have been extensively studied building upon wider DNA-based supramolecular chemistry and developing the pioneering studies of Seeman et al.64 In addition to the studies of DNA assembly an area that has received particular attention has been the use of the individual DNA bases to create surface-based self-assembled structures.65 The assembly of guanine and its derivatives on surfaces has received particular attention, in part due to interest in the formation of guanine quadruplexes that have particular importance in biological processes.66 It is notable that the adsorption of guanine onto a Au(111) surface under UHV conditions leads to the formation of guanine quartets,67 the quartets, formed by Hoogsteen-style interactions, are associated through further N–H⋯N hydrogen bonds to give rise to two-dimensional supramolecular structures.
N9-alkyl substituted guanine motifs have also been investigated but at the solid-solution interface rather than in UHV.68,69 Thus C18H37-N9-guanine has been demonstrated to form hydrogen-bonded guanine ribbons flanked by interdigitated alkyl chains on an HOPG surface. This arrangement gives rise to a two-dimensional array stabilized by both hydrogen bonding and by physisorbed alkyl chains (Fig. 8).68 Addition of 100 equivalents of potassium picrate to the solution leads to a rearrangement of the guanine ribbons to form guanine quartets with a potassium cation sitting in the central cavity of the quartet (Fig. 8). The guanine quartet can be destabilized by removal of the potassium cation by addition of the [2.2.2]cryptand sequestering agent resulting in the reassembly of the ribbon structure. The guanine quartet can be regenerated by addition of trifluoromethanesulfonic acid to the solution, which liberates the potassium cations from the [2.2.2]cryptand allowing the potassium cations to re-template the formation of the quartet structure. The interconversion of the guanine hydrogen-bonded motifs represents an elegant example of dynamic supramolecular chemistry but importantly the demonstration of the supramolecular chemistry is further enhanced by the molecular resolution of STM imaging.
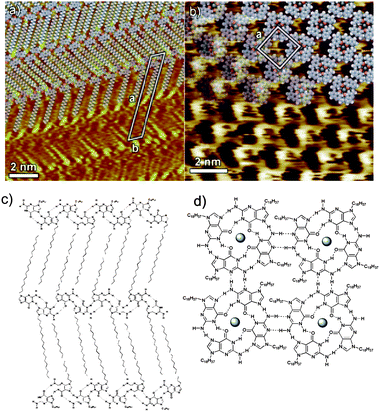 | ||
| Fig. 8 a) and c) Ribbon guanine structure formed by C18H37-N9-guanine, deposited onto an HOPG surface from a 1,2,4-trichlorobenzene solution. Addition of potassium cations leads to the formation of guanine quartet structures, b) and d).68 Reproduced with permission from Wiley-VCH Verlag GmbH and Co. KGaA. | ||
Conclusions
It can be clearly seen that surface-based supramolecular chemistry has a rich future. Many of the approaches that are familiar to supramolecular chemists who work in either the solution phase or solid state are applicable to surface-based processes. A wide variety of self-assembled structures can be readily prepared and characterised, and host–guest chemistry is also viable in the surface environment facilitating many avenues of research for the preparation of nanoscale devices.Surface-based supramolecular chemistry also offers significant differences in comparison to more traditional areas of the field. The role of the surface represents not only a challenge to interpret but also to exploit in designing novel self-assembling systems. Perhaps one of the most intriguing features of the field is the use of molecular resolution imaging (STM) to probe and characterise the self-assembled systems. The use of this technique allows unprecedented insight into the nature of the self-assembly process and perhaps more than any other approach gives a greater understanding of the reliability of self-assembly. Simple inspection of STM images of supramolecular systems reveals much greater appreciation of the complexity of systems and the competing pathways that commonly exist in the self-assembly process. Interestingly, molecular resolution also allows appreciation of systems that would be daunting to unravel using any other approach. The potential of the approach is amply demonstrated by the random molecular frameworks formed by terphenyl-3,3′′,5,5′′-tetracarboxylic acid27,28 where the molecular resolution of STM provides a level of detail and understanding of the random system that would not be feasible using more traditional methodologies. The advantages and previously undiscovered avenues of research that surface-based supramolecular chemistry facilitates can only mean that the area has a rich and highly fruitful future.
Acknowledgements
We thank the UK Engineering and Physical Sciences Research Council for financial support for our research highlighted herein. N.R.C. acknowledges the receipt of a Royal Society Leverhulme Trust Senior Research Fellowship.References
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763 CrossRef CAS
.
- D. N. Reinhoudt and M. Crego-Calama, Science, 2002, 295, 2403 CrossRef CAS
- C. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22 RSC
- L. Bartels, Nat. Chem., 2010, 2, 87 CrossRef CAS
- D. Bonifazi, S. Mohnani and A. Llanes-Pallas, Chem.–Eur. J., 2009, 15, 7004 CrossRef CAS
- K. E. Plass, A. L. Grzesiak and A. J. Matzger, Acc. Chem. Res., 2007, 40, 287 CrossRef CAS
- S. De Feyter, A. Gesquière, M. M. Abdel-Mottaleb, P. C. M. Grim and F. C. De Schryver, Acc. Chem. Res., 2000, 33, 520 CrossRef CAS
- S. De Feyter and F. C. De Schryver, Chem. Soc. Rev., 2003, 32, 139 RSC
- T. Kudernac, S. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402 RSC
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805 RSC
- C. Haensch, S. Hoeppener and U. S. Schubert, Chem. Soc. Rev., 2010, 39, 2323–2334 RSC
- Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. Northrop, H.-R. Tseng, J. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. Solares, W. A. Goddard III, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745 CrossRef CAS
- M. Surin, P. Samorì, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Angew. Chem., Int. Ed., 2007, 46, 245 CrossRef CAS
- T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619 CAS
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604 CrossRef CAS
- G. Schull, L. Douillard, C. Fiorini-Debuisschert and F. Charra, Nano Lett., 2006, 6, 1360 CrossRef CAS
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546 CrossRef CAS
- S. Narasimhan and D. Vanderbilt, Phys. Rev. Lett., 1992, 69, 1564 CrossRef CAS
- M. Ruben, J. M. Lehn and P. Muller, Chem. Soc. Rev., 2006, 35, 1056 RSC
- O. Ivasenko and D. F. Perepichka, Chem.
Soc. Rev., 2011, 40, 191–206 RSC
- S. B. Lei, C. Wang, S. X. Yin, H. N. Wang, F. Wi, H. W. Liu, B. Xu, L. J. Wan and C. L. Bai, J. Phys. Chem. B, 2001, 105, 10838 CrossRef CAS
- S. De Feyter, A. Gesquière, P. C. M. Grim, F. C. De Schryver, S. Valiyaveettil, C. Meiners, M. Sieffert and K. Müllen, Langmuir, 1999, 15, 2817–2822 CrossRef CAS
- S. Griessl, M. Lackinger, M. Edelwirth, M. Hietschold and W. M. Heckl, Single Mol., 2002, 3, 25 CrossRef CAS
- R. Gutzler, T. Sirtl, J. F. Dienstmaier, K. Mahata, W. M. Heckl, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2010, 132, 5084 CrossRef CAS
- H. Zhou, H. Dang, J.-H. Yi, A. Nanci, A. Rochefort and J. D. Wuest, J. Am. Chem. Soc., 2007, 129, 13774–13775 CrossRef CAS
- M. Blunt, X. Lin, M. C. Gimenez-Lopez, M. Schröder, N. R. Champness and P. H. Beton, Chem. Commun., 2008, 2304 RSC
- M. O. Blunt, J. Russell, M. C. Giménez-López, J. P. Garrahan, X. Lin, M. Schröder, N. R. Champness and P. H. Beton, Science, 2008, 322, 1077 CrossRef CAS
- M. O. Blunt, J. C. Russell, M. C. Gimenez-Lopez, N. Taleb, X. Lin, M. Schröder, N. R. Champness and P. H. Beton, Nat. Chem., 2011, 3, 74–78 Search PubMed
- M. Li, K. Deng, S.-B. Lei, Y.-L. Yang, T.-S. Wang, Y.-T. Shen, C. Wang, Q.-D. Zeng and C. Wang, Angew. Chem., Int. Ed., 2008, 47, 6717 CrossRef CAS
- M. Fujita, S. Nagao and K. Ogura, J. Am. Chem. Soc., 1995, 117, 1649–1650 CrossRef CAS
- F. H. Allen, W. D. S. Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25–34 RSC
- J. A. Zerkowski, J. P. Mathias and G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 4305 CrossRef CAS
- M. Mascal, J. Hansen, P. S. Fallon, A. J. Blake, B. R. Heywood, M. H. Moore and J. P. Turkenburg, Chem.–Eur. J., 1999, 5, 381 CrossRef CAS
- J.-M. Lehn, M. Mascal, A. DeCian and J. Fischer, Chem. Commun., 1990, 479 RSC
- J. A. Zerkowski, C. T. Seto and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 5473 CrossRef CAS
- M. A. Mateos-Timoneda, J. M. C. A. Kerckhoffs, M. Crego-Calama and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2005, 44, 3248 CrossRef CAS
- T. M. Bohanon, P.-L. Caruso, S. Denzinger, R. Fink, D. Mobius, W. Paulus, J. A. Preece, H. Ringsdorf and D. Schollmeyer, Langmuir, 1999, 15, 174 CrossRef CAS
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029 CrossRef CAS
- P. A. Staniec, L. M. A. Perdigão, A. Saywell, N. R. Champness and P. H. Beton, ChemPhysChem, 2007, 8, 2177 CrossRef CAS
- A. Saywell, G. Magnano, C. J. Satterley, L. M. A. Perdigão, N. R. Champness, P. H. Beton and J. N. O'Shea, J. Phys. Chem. C, 2008, 112, 7706–7709 CrossRef CAS
- J. A. Theobald, N. S. Oxtoby, N. R. Champness, P. H. Beton and T. J. S. Dennis, Langmuir, 2005, 21, 2038 CrossRef CAS
- A. Saywell, G. Magnano, C. J. Satterley, L. M. A. Perdigão, A. J. Britton, N. Taleb, M. C. Giménez-López, N. R. Champness, J. N. O'Shea and P. H. Beton, Nat. Commun., 2010, 1, 75 Search PubMed
- L. M. A. Perdigão, E. W. Perkins, J. Ma, P. A. Staniec, B. L. Rogers, N. R. Champness and P. H. Beton, J. Phys. Chem. B, 2006, 110, 12539 CrossRef CAS
- F. Silly, A. Q. Shaw, K. Porfyrakis, J. H. Warner, A. A. R. Watt, M. R. Castell, H. Umemoto, T. Akachi, H. Shinohara and G. A. D. Briggs, Chem. Commun., 2008, 4616 RSC
- L. M. A. Perdigão, P. A. Staniec, N. R. Champness and P. H. Beton, Langmuir, 2009, 25, 2278 Search PubMed
- L. M. A. Perdigão, A. Saywell, G. N. Fontes, P. A. Staniec, G. Goretzki, A. G. Phillips, N. R. Champness and P. H. Beton, Chem.–Eur. J., 2008, 14, 7600 CrossRef CAS
- A. G. Phillips, L. M. A. Perdigão, P. H. Beton and N. R. Champness, Chem. Commun., 2010, 46, 2775 RSC
- R. Madueno, M. T. Räisänen, C. Silien and M. Buck, Nature, 2008, 454, 618 CrossRef CAS
- C. Silien, M. T. Räisänen and M. Buck, Small, 2010, 6, 391 Search PubMed
- C. Silien, M. T. Raisanen and M. Buck, Angew. Chem., Int. Ed., 2009, 48, 3349 CrossRef CAS
- J. Coraux, A. T. N'Diaye, C. Busse and T. Michely, Nano Lett., 2008, 8, 565 CrossRef CAS
- S. Berner, M. Corso, R. Widmer, O. Groening, R. Laskowski, P. Blaha, K. Schwarz, A. Goriachko, H. Over, S. Gsell, M. Schreck, H. Sachdev, T. Greber and J. Osterwalder, Angew. Chem., Int. Ed., 2007, 46, 5115 CrossRef CAS
- F. Nolde, W. Pisula, S. Müller, C. Kohl and K. Müllen, Chem. Mater., 2006, 18, 3715 CrossRef CAS
- A. J. Pollard, E. W. Perkins, N. A. Smith, A. Saywell, G. Goretzki, A. G. Phillips, S. P. Argent, H. Sachdev, F. Müller, S. Hüfner, S. Gsell, M. Fischer, M. Schreck, J. Osterwalder, T. Greber, S. Berner, N. R. Champness and P. H. Beton, Angew. Chem., Int. Ed., 2010, 49, 1794 Search PubMed
- N. Berdunov, A. J. Pollard and P. H. Beton, Appl. Phys. Lett., 2009, 94, 043110 Search PubMed
- G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. N. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37 CrossRef CAS
- L. M. A. Perdigão, N. R. Champness and P. H. Beton, Chem. Commun., 2006, 538 RSC
- P. A. Staniec, L. M. A. Perdigão, B. L. Rogers, N. R. Champness and P. H. Beton, J. Phys. Chem. C, 2007, 111, 886–893 CrossRef CAS
- D. L. Keeling, N. S. Oxtoby, C. Wilson, M. J. Humphry, N. R. Champness and P. H. Beton, Nano Lett., 2003, 3, 9 CrossRef CAS
- L. M. A. Perdigão, G. N. Fontes, B. L. Rogers, N. S. Oxtoby, G. Goretzki, N. R. Champness and P. H. Beton, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 245402 CrossRef
- C.-A. Palma, J. Bjork, M. Bonini, M. S. Dyer, A. Llanes-Pallas, D. Bonifazi, M. Persson and P. Samorì, J. Am. Chem. Soc., 2009, 131, 13062 CrossRef CAS
- L. Piot, C.-A. Palma, A. Llanes-Pallas, M. Prato, Z. Szekrényes, K. Kamarás, D. Bonifazi and P. Samorì, Adv. Funct. Mater., 2009, 19, 1207 Search PubMed
- M. Stoehr, M. Wahl, C. H. Galka, T. Riehm, T. A. Jung and L. H. Gade, Angew. Chem., Int. Ed., 2005, 44, 7394 CrossRef CAS
- N. C. Seeman, Nature, 2003, 421, 427 CrossRef
- Q. Chen and N. V. Richardson, Nat. Mater., 2003, 2, 324 CrossRef CAS
-
S. Neidle, S. Balasubramanian, Quadruplex nucleic acids, Royal Society of Chemistry, Cambridge, 2006 Search PubMed
- R. Otero, M. Schock, L. M. Molina, E. Laegsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Angew. Chem. Int. Ed, 2005, 44, 2270 CrossRef CAS
- A. Ciesielski, S. Lena, S. Masiero, G. Piero Spada and P. Samorì, Angew. Chem. Int. Ed, 2010, 49, 1963 CAS
- A. Ciesielski, R. Perone, S. Pieraccini, G. Piero Spada and P. Samorì, Chem. Commun., 2010, 46, 4493 RSC
| This journal is © The Royal Society of Chemistry 2011 |