Molecular nature of support effects in single-site heterogeneous catalysts: silicavs.alumina
Fernando
Rascón
,
Raphael
Wischert
and
Christophe
Copéret
*
ETH Zürich, Department of Chemistry, Laboratory of Inorganic Chemistry, Wolfgang Pauli Strasse 10, CH-8093, Zürich, Switzerland. E-mail: ccoperet@inorg.chem.ethz.ch; Tel: +41-44-633 9394
First published on 12th May 2011
Abstract
Well-defined silica- and alumina-supported catalysts can display very different reactivity, which is often attributed to support effects. Advanced spectroscopic studies in combination with computational chemistry show that the origin of these differences is mainly molecular in nature. Silica partially dehydroxylated at 700 °C features mainly isolated silanols (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiOH) as surface functional groups, which can be exploited to generate well-defined and site isolated siloxy metal species. In contrast, alumina, a more complex support, exhibits both hydroxyl groups and highly reactive Al,O Lewis acid and base surface sites. Therefore, it is better understood as a multifunctional and adaptable ligand, which can lead to a variety of surface species, often more reactive (cationic) and/or stable than their known silica counterparts.
SiOH) as surface functional groups, which can be exploited to generate well-defined and site isolated siloxy metal species. In contrast, alumina, a more complex support, exhibits both hydroxyl groups and highly reactive Al,O Lewis acid and base surface sites. Therefore, it is better understood as a multifunctional and adaptable ligand, which can lead to a variety of surface species, often more reactive (cationic) and/or stable than their known silica counterparts.
 Fernando Rascón, Christophe Copéret and Raphael Wischert | Dr. Fernando Rascón (left), Prof. Christophe Copéret (center) and Dr. Raphael Wischert (right) recently moved to the Department of Chemistry (Institute of Inorganic chemistry) of ETH Zürich (Switzerland) from C2P2/CPE Lyon (France). The group’s research interests lie in the molecular understanding and rational development (structure-property relationship) of heterogeneous catalysts and functional materials. Thus, besides evaluation of properties (e.g. catalytic performances), the research group is involved in material chemistry, controlled surface chemistry, and characterization of surface species and reaction intermediates using the combination of spectroscopy and computational chemistry. For more information, see: http://www.coperetgroup.ethz.ch/. |
Introduction
The support plays a protagonist role in heterogeneous catalysis, including the class of so-called single-site catalysts.1 For instance, clear differences in catalytic performance in terms of activity, selectivity and stability have been reported for supported silicavs.alumina single-site catalysts for various reactions, such as alkene polymerisation,2hydrogenation,3 and metathesis.4 Our aim is to discuss the support effects at a molecular level, focusing on selected examples of these systems. This implies not only describing surface functionalities as ligands of the grafted organometallic entities—as in molecular organometallic chemistry—but also including the effect of the presence or absence of (i) interactions between the surface and the grafted molecular entities or the substrate, (ii) Lewis acidic sites, or “defects”. All of these factors can play a major role on the structure of surface species, whether being pre-catalyst (surface complexes) or reaction intermediates. Because of their effect on the catalytic performance, these factors should be taken into account when designing heterogeneous catalysts.Silica-supported systems
Generalities on silica
Silica is probably one of the most studied oxide supports; it is an amorphous solid composed of a network of tetrahedral [SiO4]4− units, and it can develop large specific surface areas (100–1000 m2 g−1).5 Crystalline silicas also exist but they are not used as catalyst supports because of their small surface area (few m2 g−1). The surface of amorphous silica surfaces is composed of siloxane bridges (Si–O–Si) and silanol groups (SiOH), whose concentration and types (isolated, geminal or vicinal, Fig. 1a) depend on the temperature of treatment under vacuum or gas flow.5b,5d Upon thermal treatment, the overall density of silanol groups decreases nearly exponentially with temperature by self-condensation (Fig. 1b).5e This dehydroxylation process yields water and siloxane bridges of decreasing annularity, and it is accompanied by an increased ratio of isolated silanols.
 | ||
| Fig. 1 a) Types of silanols. b) Effect of temperature on surface-OH coverage (θOH) and infrared spectra of silica partially dehydroxylated at 200 (SiO2-(200)) and 700 °C (SiO2-(700)). c) Types of siloxane bridges. | ||
Partial dehydroxylation takes place without significant loss of specific surface area for temperatures up to around 700 °C for Aerosil® silica. At this temperature, this solid—referred to as SiO2-(700)—presents a statistical population of isolated silanols with a coverage of ca. 0.8 OH m−2. Above this temperature, the surface area starts to drop significantly, and reactive, strained siloxane bridges are formed (Fig. 1c). Thermal treatment on mesoporous silica such as MCM-41 can lead to structural collapse, resulting in a sharp loss of specific surface area, if it is performed too fast or at temperatures above 500–600 °C.5a Thereafter, we will mainly discuss the surface chemistry of Aerosil® silica (200 m2 g−1), a support composed of pure silica nanoparticles having a diameter of ca.12 nm. Nonetheless, such discussion can be easily transposed to mesoporous silica, minor differences being the OH density (within 20%), the stability of the material and the roughness of the surface.6
Effect on the structure of surface complexes
With a low density of isolated silanols, SiO2-(700) has been the support of choice for generating well-defined complexes attached by a single covalent bond to the surface (vide infra).1a,7 In most cases, grafting perhydrocarbyl transition-metal complexes8 on SiO2-(700) leads to the formation of an alkane and a surface species in which one of the M–C bonds has been replaced by a M–O bond without otherwise significantly modifying the structure of the metal fragment (Fig. 2a,b).9 Lower dehydroxylation temperatures provide supports with a higher density of vicinal silanols, leading to a higher amount of bis-grafted species; the highest being obtained for silica treated at temperatures of ca. 200 °C.2a,10 Grafting is not limited to perhydrocarbyl complexes,11 since alkoxides12 and amides13 can also be used in the same way to access well-defined surface species (Fig. 2c, X = OR or NR2);10a,14 here an alcohol and an amine are formed and released, but can be easily removed from the rather hydrophobic surface of dehydroxylated silica by washing.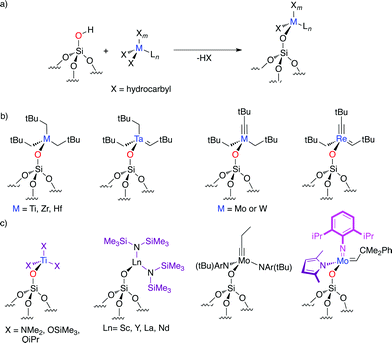 | ||
| Fig. 2 a) Grafting of an organometallic compound onto a silica surface. b) Perhydrocarbyl silica-supported surface species. c) Examples of amido- and alkoxy-based surface species. | ||
A more detailed understanding of silica supported systems can be illustrated with [(SiO)Re(CtBu)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHtBu)(CH2tBu)] (Fig. 3a), a well-defined surface complex, fully characterized by IR, solid-state NMR and EXAFS, and selectively obtained from the grafting of [Re(CtBu)(CHtBu)(CH2tBu)2] on SiO2-(700).9a,15 Formed selectively as the syn-isomer, the resulting surface complex displays a C–H agostic interaction for its alkylidene ligand, as evidenced by the low JC–H coupling constant. Further thermal or photochemical treatment of the complex leads to a mixture of syn and anti isomers,15,16 the latter presenting no agostic interaction. Grafting of isoelectronic and isostructural Mo- and W-imido complexes also selectively generates the corresponding syn-isomer (Fig. 3b, X = CH2tBu and E = NAr).17
CHtBu)(CH2tBu)] (Fig. 3a), a well-defined surface complex, fully characterized by IR, solid-state NMR and EXAFS, and selectively obtained from the grafting of [Re(CtBu)(CHtBu)(CH2tBu)2] on SiO2-(700).9a,15 Formed selectively as the syn-isomer, the resulting surface complex displays a C–H agostic interaction for its alkylidene ligand, as evidenced by the low JC–H coupling constant. Further thermal or photochemical treatment of the complex leads to a mixture of syn and anti isomers,15,16 the latter presenting no agostic interaction. Grafting of isoelectronic and isostructural Mo- and W-imido complexes also selectively generates the corresponding syn-isomer (Fig. 3b, X = CH2tBu and E = NAr).17
![a) Thermal interconversion of [(SiO)Re(CtBu)(CHtBu)(CH2tBu)] into its syn/anti isomers. b) Representation of d0 ML4 alkylidyne/alkylidene supported olefin metathesis catalysts (M = Mo, W, Re). c) Disposition of the ligands for d0 ML4 complexes.](/image/article/2011/SC/c1sc00073j/c1sc00073j-f3.gif) | ||
| Fig. 3 a) Thermal interconversion of [(SiO)Re(CtBu)(CHtBu)(CH2tBu)] into its syn/anti isomers. b) Representation of d0 ML4 alkylidyne/alkylidene supported olefin metathesis catalysts (M = Mo, W, Re). c) Disposition of the ligands for d0 ML4 complexes. | ||
Computational studies using various models of siloxy ligands [H3SiO–, POSS– (POSS– = (c-C5H9)7Si7O12SiO–), and SiO– of cristobalite models] show that the syn isomer is thermodynamically more stable and presents an agostic interaction resulting from the interaction of the σ(C–H) orbital of the alkylidene ligand with the antibonding σ*(ME) orbital of the alkylidyne ligand.18 It is noteworthy that the simplest possible model of a silica surface, based on a H3SiO– fragment, reproduces quite well the essential properties of the actual system (structure, syn/anti-ratio and spectroscopic data), which shows that the anchoring point on a silica surface can be viewed just as a molecular silanol ligand.18b
Finally, for d0 alkylidene complexes (Fig. 3b), the substitution of one of the neopentyl ligands by a siloxy provokes a change in their structure from an almost perfect tetrahedron into a distorted one, with an opening of the face trans to the remaining neopentyl ligand X ([OEC face], Fig. 3c). This is in line again with the weaker σ-donor character of the siloxy ligand compared to alkyl or other ligands with more electronegative atoms, e.g. N from amido. In fact, this is observed for all d0 ML4 alkylidene complexes having dissymmetric ligand sets (X = CH2tBu, NR2).19 The nature of the siloxy ligand (a weak σ-donor) can be clearly illustrated by the change of geometry and structure upon grafting of Os(CHtBu)2(CH2tBu)2—a d2ML4 tetrahedral complex—on SiO2-(700), yielding (SiO)Os(CtBu)(CH2tBu)2.20 This surface complex adopts a see-saw structure and bears one alkylidyne and two alkyl ligands (Fig. 4).
 | ||
| Fig. 4 a) Grafting of Os(CHtBu)2(CH2tBu)2 onto SiO2-(700). b) Optimised geometry of (SiO)Os(CtBu)(CH2tBu)2 on a periodic surface of silica. | ||
Effects on the reactivity
In numerous Lewis acid-catalyzed reactions such as alkene oxidation21 as well as alkene/alkyne metathesis,4,22silica-grafted species based on lanthanides, group 3 or d0 transition-metal complexes often display higher catalytic performance (activity and stability) than their molecular counterparts.9d, 14b,23 A simple explanation involves the formation of mononuclear and more electrophilic (more Lewis acidic) centres upon including a siloxy rather than an alkoxide ligand. In the particular case of silica-supported alkylidene complexes, theoretical studies have allowed us to delineate the origin of the reactivity in alkene metathesis.19,24 Here, the siloxy ligand (a weak σ-donor ligand) sensibly affects the structure and the stability of reaction intermediates and transition states (Fig. 5a). The key step in metathesis is the coordination of the incoming alkene, which requires the distortion of the almost tetrahedral species in order to form the π-alkene complex having a TBP geometry. The approach can only take place from one of the two faces presenting the alkylidene ligand in the proper orientation to form the metallacycle intermediate (attack on [OEC] and [XEC] faces, see Fig. 3c). Overall, the coordination of the alkene through the [OEC] face is associated with the lowest energy trigonal bipyramidal (TBP) transition state (TS), because the strong σ-donor ligand (X = alkyl or amide) occupies an apical position trans to the incoming alkene, still far from the metal center. The two other strong σ-donor ligands (alkylidene and E = alkylidyne or imido) share the basal plane of the TBP with the weak σ-donor ligand (siloxy). The other approach (on the [XEC] face) is disfavored because it leads to a TS with three strong σ-donor ligands in the basal plane of the TBP. Finally, note that the preferred face of attack is also the most open one in the alkylidene precursor, thus showing that a disymmetric system prepares the catalyst, which contributes to its higher reactivity. In turn, the analysis of these ligand effects has led to a deeper understanding of the performance of alkene metathesis catalysts.19,24,25 Based on this structure–activity relationship, better performing silica-supported catalysts (Fig. 5b)4,14d,14e,26 and even homogeneous systems (Fig. 5c) have been developed.27 | ||
| Fig. 5 a) Mechanism for alkene metathesis. b) Specific role of each ligand for Mo-catalysts from structure–activity relationship. c) Example of a highly efficient dissymmetric homogeneous alkene metathesis catalyst containing a stereogenic Mo centre.27b | ||
Effects on the stability
Grafting on silica materials translates into site isolation, which brings higher stability by impeding bimolecular decomposition pathways, including self-aggregation. This has been shown in alkene metathesis by the higher stability of silica supported systems compared to their molecular equivalents (vide supra).17a Such higher stability also enables supported alkyl-alkylidene metal complexes to be used as pre-catalysts in alkane metathesis, which is performed at 150 °C.28,29 Further treatment of silica-supported perhydrocarbyl Zr and Ta-complexes with H2 yields the corresponding monomeric hydrides,30 whose molecular equivalents are not known in the absence of bulky ligands (Fig. 6). This is due to the strong M–O bond early transition metals form with the silica support. The metal centres experience additional stabilisation by the formation of bond(s) with the silica surface upon the opening of adjacent siloxane bridges. These supported hydrides readily activate the C–H bonds of unactivated alkanes at low temperature and display a reactivity unprecedented in molecular chemistry: low temperature hydrogenolysis of alkanes31 and polymers,32alkane metathesis,33 and homologation of methane.34 Supported tantalum hydrides can even cleave the N2 molecule.35 | ||
| Fig. 6 Zirconium and tantalum surface metal hydrides obtained by H2 treatment. | ||
Alumina-supported systems
Generalities on alumina
Alumina (Al2O3) is a more complex support, in spite of its higher crystallinity. In fact, depending on its preparation, which typically consists in treating aluminium hydroxide precursors at high temperature, numerous polymorphs can be formed.36α-Al2O3 (corundum), as the thermodynamically most stable phase is formed above 1000 °C and has a very low surface area (<20 m2 g−1). It is used in catalytic processes as a chemically inert support material for metals when high mechanical and thermal resistance is required (for example in the Haber–Bosch process for ammonia synthesis). Metastable aluminas formed by dehydration at 500–800 °C are known as transition or activated aluminas. They are high-surface area (80–250 m2 g−1) porous materials which find wide application as catalysts or as catalyst supports, γ-Al2O3 being the most widely used.36,37 Despite considerable efforts devoted to its study, the exact bulk structure and surface sites of γ-Al2O3 are still not fully understood and are a matter of debate.38 Extensive investigations by periodic DFT calculations in combination with experimental data have provided a more detailed structural description of the bulk of γ-Al2O339 and its surface sites.38,40 The bulk contains aluminium ions in tetrahedral (25%) and octahedral (75%) coordination. The unit cell of the most abundant (110) termination (ca. 80%)40b,41 is composed of one tricoordinated AlIII and two types of tetracoordinated AlIV sites, namely AlIVa and AlIVb (Fig. 7a), resulting from tetrahedral and octahedral bulk Al atoms, while the less abundant (100) termination (ca. 10%) exposes only AlV sites. The Lewis acidity of these low-coordinated Al atoms follows the order AlIII≫AlIVb>AlV>AlIVa. The surface also exposes oxygen atoms with Lewis basicity O2a/b > O3a/b.40b However, these sites are partially occupied by OH groups or protons (generated by dissociation of H2O on Al,O sites), even after high temperature pretreatment. The coordination of these hydroxyls (μ1, μ2 and μ3), depends on the type and number of Al sites to which they are bound. In addition, non-dissociated H2O molecules coordinated to Al sites can also be stabilized on the surface, increasing the number of hydroxyl types (Fig. 7b). In comparison to silica, the corresponding IR spectrum of the OH-region (see for example that for γ-Al2O3-(500) in Fig. 8a) is therefore rather complex, but it was nevertheless possible to fully assign the bands with the aid of DFT calculations.40 | ||
| Fig. 7 a–d) Structure of the (110) termination of γ-Al2O3: a) fully dehydroxylated surface (s0); b) covered by 3 H2O per unit cell, ca. 9 OH nm−2 (s3); c) with free AlIII site, covered by 1 H2O per unit cell, ca. 3 OH nm−2 (s1); d) with free AlIII site, covered by 2 H2O per unit cell, ca. 6 OH nm−2 (s2). Note that for the most stable surfaces, covered by 1 or 2 H2O, AlIII, the most Lewis acidic site is occupied by an OH group. Only the top two layers of the periodical slab are represented. A dashed line indicates the unit cell. Al: yellow, O originating from the γ-Al2O3 bulk: red, O originating from H2O dissociation: purple, H: white balls, AlIVb after surface reconstruction: green circle. All distances are given in Å. | ||
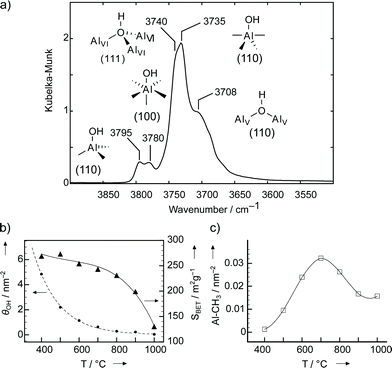 | ||
| Fig. 8 a) Infrared spectrum of γ-Al2O3 dehydroxylated at 500 °C in vacuum (1.77 × 10−3 Pa). Assignment of the OH-bands according to Digne et al.40b b–c) Influence of thermal treatment on the surface of γ-alumina: b) Surface OH coverage (θOH, dotted line); Surface area (SBET, solid line). c) Abundance of Al–CH3 (C–H activation) upon contact with CH4. Adapted from ref. 43. | ||
Here, the stabilisation of the surface upon hydration is worth mentioning. In contrast to the less abundant (100), the (110) termination is predicted to be thermodynamically unstable in its fully dehydrated form (s0).40
Upon thermal treatment the OH density (θOH) decreases exponentially with temperature,42,43 reaching 2 and 0.6 OH nm−2 at 500 and 700 °C, respectively (Fig. 8b). Lewis acid sites are formed by removal of chemisorbed H2O (dissociated and non-dissociated) by thermal treatments above 400 °C. The most reactive, tricoordinated AlIII sites (“defects” or α-sites44) are noteworthy in terms of their reactivity: i) the AlIII,O pairs stoichiometrically activate the C–H bond of methane to generate Al–CH3 and O–H surface species,33c,43,45 and ii) are therefore most probably involved in the low-temperature H/D-exchange reaction of CH4 and D2 into CH3D and HD mixtures (via C–H activation).42,46 The density of these sites increases up to about 700 °C, reaching a maximum of 0.03 nm−2, and then vanishes as the bulk gradually transforms into θ- and α-alumina, the thermodynamically more stable phases. This process is accompanied by a sharp loss of surface area (Fig. 8b).43 Therefore γ-Al2O3 is most reactive towards CH4 dissociation at an OH-coverage of ca. 0.6 OH nm−2, which strongly suggests the presence of the highly reactive AlIII. This is somewhat counterintuitive as these sites should be in principle occupied by OH groups as discussed above.47
Recent studies43 reveal that AlIII sites can indeed exist in the presence of surface hydroxylation up to relatively high OH-coverages of 6 OH nm−2 (corresponding to a pretreatment temperature of ca. 400 °C, s2, see Fig. 7d). The unexpected occurrence of a stable s2 surface with free AlIII sites is largely due to a reorganization of the surface, with Al sites originally in truncated octahedral geometry ending up upon hydration in a more stable tetrahedral coordination. However, because of the high rigidity of the s2 surface (reconstruction, coordination of AlIII by a 2nd-layer O atom), this AlIII site is unreactive towards CH4 activation, in line with the experimental findings.43,45
At lower water coverage (s1, θOH = 3 OH nm−2, Fig. 7c) the probability of finding AlIII sites is lower, however they are highly reactive towards CH4, as observed experimentally. This is due to the formation of highly reactive frustrated Lewis acid-base pairs:48 the Lewis acidity of AlIII is high on s1 and the basicity of the oxygen partner (O3a, Fig. 7d) is strongly increased by the hydration of the adjacent AlIVa sites. In summary, the hydroxylation of the surface has a dual role: firstly, as stated, it stabilizes the otherwise unstable (110) termination and thus enables AlIII sites. Second, it maximises the reactivity of these sites at an optimal water coverage, modeled by s1.
These findings illustrate the richness and complexity of the alumina surface. Like silica, alumina features OH groups but these exhibit different coordinations, in contrast to silica (unless the latter is treated at low temperature). The main difference, however, consists in the presence of Lewis acidic and basic Al and O sites. Unlike silica, where all the silicon atoms are fixed in a rather regular, tetrahedral environment and are therefore chemically inert, alumina exhibits highly reactive, Lewis acidic Al atoms thanks to its configurational flexibility. At the same time, the crucial role of oxygen basicity should be reemphasized for reactions where Al,O pairs are involved, such as the activation of C–H bonds.
Overall, γ-alumina exhibits a complex and flexible surface with a rich palette of surface sites and functionalities; it is therefore not surprising that this support exerts a profound effect on the structure, reactivity and stability of surface species involved in catalytic events, contributing to the rationale of the so-called support effect.
Effects on the structure of surface complexes
The surface hydroxyls are involved in the grafting of organometallic complexes in several cases.2b, 49 On alumina (either γ or δ) partially dehydroxylated at 500 °C, where θOH is ca. 2 OH nm−2, the grafting of Zr(CH2tBu)4 leads to the full consumption of surface OH groups and to the formation of 2 equiv of tBuCH3, yielding the bis-grafted species (AlSO)2Zr(CH2tBu)2.50 A detailed study combining IR, EXAFS, solid-state NMR spectroscopy and computational modelling shows that several surface species are present, but in particular, that cationic Zr complexes are formed, as evidenced by the formation of neopentyl aluminate (Fig. 9a).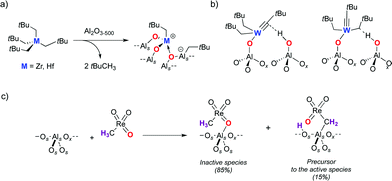 | ||
| Fig. 9 a) Grafting of M(CH2tBu)4 onto Al2O3-(500) leading to cationic species. b) Interactions of (AlSO)W(CtBu)(CH2tBu)2 with neighboring surface hydroxyls on alumina. c) Grafting of CH3ReO3 onto Al2O3-(500). | ||
This behaviour evidences the multifunctional nature of alumina surfaces. In fact, analogous cationic surface species have also been observed with other group 4 transition metal complexes, such as Cp*ZrMe351 or Hf(CH2tBu)4,52 as well as lanthanide and actinide complexes.3a, 53 In contrast, grafting of W(CtBu)(CH2tBu)3—containing a less electrophilic metal centre—leads to partial consumption of surface OH groups and to the selective grafting on AlIVOH, generating the monografted surface species (AlSO)W(CtBu)(CH2tBu)2, where the hydrocarbyl moieties interact with the remaining surface hydroxyls (Fig. 9b). Here, the formation of cationic species is not observed.50 Grafting of CH3ReO3 is also noteworthy: it does not undergo chemical reaction with OH-groups but attaches to the surface via multiple Lewis acid–base interactions involving the coordination of its oxo ligands to Al sites on the one hand and that of the Re center to surface O atoms on the other hand (Fig. 9c). Additionally, AlSCH2ReO3 species are formed via C–H activation of the methyl ligand of CH3ReO3 on Al,O sites.54
Effects on the reactivity
As stated in the introduction, major differences in reactivity have been found for silicavs.alumina supported single-site catalysts. In the field of polymerisation, in particular for catalysts based on d0 early transition-metals, the major differences arise from the fact that supported systems on silica generate neutral species, while cationic species are formed on alumina (vide supra). The same is probably true for the hydrogenation catalysts, based on actinide, lanthanide, group 3 or early transition-metals.3a, 51a, 53Similar strong support effects are known for Re-based catalysts in the field of alkene metathesis. While Re2O7/SiO2 is unreactive, one of the best performing systems is Re2O7/γ-Al2O3,55 which even becomes tolerant to functionalized alkenes such as methyl oleate when Me4Sn is used as an additive.56 Despite years of research,55a poor molecular insight has emerged. In fact, the key alkylidene species which would explain the olefin metathesis activity has never been observed, and its formation from the ReO species remains obscure. However, accumulated evidence suggests that it could be generated through a pseudo-Wittig reaction.57 In particular, the origin of the reactivity of Re2O7/γ-Al2O3 has been linked to the Lewis acidic properties of γ-alumina,55 but so far it has been difficult to obtain a more detailed understanding of the actual structure of the active sites or of the ultimate role of the alkyl tin additive. One of the standing difficulties studying this catalytic system is the low content of active sites, accounting for only 2% of the surface Re centres.57d, 58 This corresponds to a density of ca. 0.04 active sites per nm2, a value coinciding with the number of “defect” sites present on γ-alumina partially dehydroxylated at 500 °C (0.03 nm−2, vide supra). While possibly fortuitous, such correspondence suggests the implication of “defects” for the generation of the active sites. In this respect, studies on a related model system, CH3ReO3 supported on γ-Al2O3 dehydroxylated at 500 °C (CH3ReO3/γ-Al2O3),54,59 are insightful, as both systems possess the same selectivity in 2-butene metathesis at low contact times (Z/E = 0.4) and thus possibly similar active sites.59c For CH3ReO3/γ-Al2O3 it was shown that the active sites—or more precisely—the precursor and resting state of the active site is (Als)CH2ReO3, formed by the C–H activation of CH3ReO3 on reactive Al,O pairs upon grafting (Fig. 9c).54,59c It is noteworthy that CH3ReO3/γ-Al2O3, where the key alkylidene ligand is already in place in a masked form as (Als)CH2ReO3, does not require activating agents to be tolerant to functionalized alkenes.59a This points to the role of Me4Sn in the metathesis of functionalized alkenes with Re2O7/γ-Al2O3, viz generating the alkylidene ligand. In the absence of Me4Sn, Re2O7/γ-Al2O3 is inactive with such polar substrates because of the poisoning of the necessary Lewis acid sites by the ester group—a Lewis base. These findings suggest that the beneficial effect of additives like Me4Sn on Re2O7/γ-Al2O3 arises from the in situ formation and subsequent grafting of CH3ReO3 or species alike. Indeed, CH3ReO3 can be efficiently prepared by the reaction of Me4Sn with Re2O7.60 The systems CH3ReO3/γ-Al2O3 and Re2O7/γ-Al2O3 stand as examples of how catalytic activity originates from the “defect” sites of the support.
Effects on the stability
While silica and alumina-supported tantalum alkane metathesis catalysts do not display substantial differences in terms of support effects, tungsten-based systems are a noteworthy example, where the alumina-supported systems outperform their silica-supported counterparts, the latter being almost inactive.61 Here, the difference is mainly due to a higher stability of the active sites. This is particularly evident during the synthesis of silica-supported tungsten hydrides, for which only a small fraction of the (SiO)W(CtBu)(CH2tBu)2 species is converted into the corresponding hydrides on silica because of sintering and formation of aggregates, while rather well-defined tungsten hydrides are generated from the corresponding isostructural alumina supported system, (AlsO)W(CtBu)(CH2tBu)2. Such difference has been explained by the stabilization of tungsten oxo species by adjacent Lewis acidic sites.33b,62 This shows that Lewis acidic sites do not only provide a way to activate metal centers but can also participate in stabilizing reactive species.
Conclusions and perspectives
In the selected examples presented, we have shown the molecular nature of the interaction of surface functionalities of oxides, namely silica and alumina, with metal centers. These interactions can lead to highly reactive, yet stable species,63 whose catalytic performance can surpass those found in molecular chemistry. While partially dehydroxylated silica can be viewed in a simplified way as a molecular siloxy ligand (SiOH) with the advantage of site isolation, alumina exhibits a more complex surface chemistry because of its multifunctional nature by the presence of Brønsted (OH) and Lewis (Al) acidic as well as Lewis basic (O) sites. These diverse environments lead to a variety of effects ranging from the formation of unusual species (cationic centres in group 4 polymerisation catalysts and masked carbenes “Als(CH2)ReO3” in CH3ReO3/γ-Al2O3) to the stabilization of surface complexes (e.g.Group 4–6 metal hydrides) with unprecedented reactivity. We have demonstrated that understanding supported catalysts requires a deep knowledge of the surface at a molecular level. Indeed, many heterogeneous catalysts display support effects, but their origin remains poorly understood. Considering the complexity of heterogeneous catalysts, it is clear that future research efforts will have to involve the development of yet more advanced spectroscopic techniques (in situ/operando) in combination with computational chemistry, along with detailed kinetic investigations and more controlled preparation processes. The better understanding of surfaces and interfaces in catalytic systems is crucial for the rational development of catalytic materials, which are the key for sustainable development.
References
- (a) C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156 CrossRef CAS; (b) J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456 CrossRef CAS; (c) M. Tada and Y. Iwasawa, Coord. Chem. Rev., 2007, 251, 2702 CrossRef CAS.
- (a) D. G. H. Ballard, Adv. Catal., 1973, 23, 263 CAS; (b) D. G. H. Ballard, Coord. Polym., 1975, 223 Search PubMed; (c) Y. I. Yermakov, B. N. Kuznetsov and V. A. Zakharov, Stud. Surf. Sci. Catal., 1981, 8, 59 Search PubMed.
- (a) T. J. Marks, Acc. Chem. Res., 1992, 25, 57 CrossRef CAS; (b) C. Copéret, in The Handbook of Homogeneous Hydrogenation, ed. Johannes G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2008, 111–151 Search PubMed.
- C. Copéret, Dalton Trans., 2007, 5498 RSC.
- (a) A. P. Legrand, The Surface Properties of Silica, Wiley, New York, 1998 Search PubMed; (b) B. A. Morrow, in Stud. Surf. Sci. Catal., ed. J. L. G. Fierro, Elsevier, 1990, vol. 57, Part 1, A161–A224 Search PubMed; (c) C. Setzer, G. van Essche and N. Pryor, in Handbook of Porous Solids, ed. F. Schüth, K. Sing and J. Weitkamp, Wiley-VCH, Weinheim, 2008, 1543–1591 Search PubMed; (d) B. A. Morrow and A. J. Mcfarlan, J. Non-Cryst. Solids, 1990, 120, 61 CrossRef CAS; (e) L. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1 CrossRef CAS.
- (a) I. G. Shenderovich, G. Buntkowsky, A. Schreiber, E. Gedat, S. Sharif, J. Albrecht, N. S. Golubev, G. H. Findenegg and H. H. Limbach, J. Phys. Chem. B, 2003, 107, 11924 CrossRef CAS; (b) J. Gath, G. L. Hoaston, R. L. Vold, R. Berthoud, C. Copéret, M. Grellier, S. Sabo-Etienne, A. Lesage and L. Emsley, Phys. Chem. Chem. Phys., 2009, 11, 6962 RSC.
- J.-M. Basset, J. P. Candy and C. Copéret, in Comprehensive Organometallic Chemistry III, ed. H. C. Robert and D. M. P. Mingos, Elsevier, Oxford, 2007, 499–553 Search PubMed.
- (a) R. R. Schrock and G. W. Parshall, Chem. Rev., 1976, 76, 243 CrossRef CAS; (b) R. R. Schrock, Acc. Chem. Res., 1979, 12, 98 CrossRef CAS; (c) R. R. Schrock, Acc. Chem. Res., 1986, 19, 342 CrossRef CAS.
- (a) M. Chabanas, A. Baudouin, C. Copéret and J.-M. Basset, J. Am. Chem. Soc., 2001, 123, 2062 CrossRef CAS; (b) R. Petroff Saint-Arroman, M. Chabanas, A. Baudouin, C. Copéret, J.-M. Basset, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2001, 123, 3820 CrossRef CAS; (c) M. Chabanas, E. A. Quadrelli, B. Fenet, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage and L. Emsley, Angew. Chem., Int. Ed., 2001, 40, 4493 CrossRef CAS; (d) R. Petroff Saint-Arroman, J.-M. Basset, F. Lefebvre and B. Didillon, Appl. Catal., A, 2005, 290, 181 Search PubMed; (e) E. Le Roux, M. Taoufik, M. Chabanas, D. Alcor, A. Baudouin, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage, S. Hediger and L. Emsley, Organometallics, 2005, 24, 4274 CrossRef.
- (a) A. O. Bouh, G. L. Rice and S. L. Scott, J. Am. Chem. Soc., 1999, 121, 7201 CrossRef CAS; (b) L. Lefort, M. Chabanas, O. Maury, D. Meunier, C. Copéret, J. Thivolle-Cazat and J.-M. Basset, J. Organomet. Chem., 2000, 593, 96 Search PubMed.
- (a) R. R. Schrock, Chem. Rev., 2002, 102, 145 CrossRef CAS; (b) R. R. Schrock, Chem. Rev., 2009, 109, 3211 CrossRef CAS.
- N. Y. Turova, E. P. Turevskaya, V. G. Kessler and M. I. Yanovskaya, The Chemistry of Metal Alkoxides, Springer, US, 2002 Search PubMed.
- M. Lappert, A. Protchenko, P. Power and A. Seeber, Metal Amide Chemistry, John Wiley & Sons, Ltd, Chinchester, UK, 2008 Search PubMed.
- (a) C. Roveda, T. L. Church, H. Alper and S. L. Scott, Chem. Mater., 2000, 12, 857 Search PubMed; (b) J. Jarupatrakorn and T. D. Tilley, J. Am. Chem. Soc., 2002, 124, 8380 CrossRef CAS; (c) H. Weissman, K. N. Plunkett and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 585 CrossRef CAS; (d) F. Blanc, J. Thivolle-Cazat, J.-M. Basset, C. Copéret, A. S. Hock, Z. J. Tonzetich, A. Sinha and R. R. Schrock, J. Am. Chem. Soc., 2007, 129, 1044 CrossRef CAS; (e) F. Blanc, R. Berthoud, A. Salameh, J.-M. Basset, C. Copéret, R. Singh and R. R. Schrock, J. Am. Chem. Soc., 2007, 129, 8434 CrossRef CAS; (f) F. Blanc, R. Berthoud, C. Copéret, A. Lesage, L. Emsley, R. Singh, T. Kreickmann and R. R. Schrock, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12123 Search PubMed.
- M. Chabanas, A. Baudouin, C. Copéret, J.-M. Basset, W. Lukens, A. Lesage, S. Hediger and L. Emsley, J. Am. Chem. Soc., 2003, 125, 492 CrossRef CAS.
- A. Lesage, L. Emsley, M. Chabanas, C. Copéret and J.-M. Basset, Angew. Chem., Int. Ed., 2002, 41, 4535 CrossRef CAS.
- (a) F. Blanc, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage, L. Emsley, A. Sinha and R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 1216 CrossRef CAS; (b) B. Rhers, A. Salameh, A. Baudouin, E. A. Quadrelli, M. Taoufik, C. Coperet, F. Lefebvre, J.-M. Basset, X. Solans-Monfort, O. Eisenstein, W. W. Lukens, L. P. H. Lopez, A. Sinha and R. R. Schrock, Organometallics, 2006, 25, 3554 CrossRef CAS.
- (a) X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, Organometallics, 2005, 24, 1586 CrossRef CAS; (b) X. Solans-Monfort, J. S. Filhol, C. Copéret and O. Eisenstein, New J. Chem., 2006, 30, 842 RSC; (c) A. Poater, X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, Dalton Trans., 2006, 3077 RSC.
- X. Solans-Monfort, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2010, 132, 7750 CrossRef CAS.
- R. Berthoud, N. Rendon, F. Blanc, X. Solans-Monfort, C. Copéret and O. Eisenstein, Dalton Trans., 2009, 5879 RSC.
- S. T. Oyama, in Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, ed. S. T. Oyama, Elsevier, Amsterdam, 2008, 3–99 Search PubMed.
- W. Zhang and J. S. Moore, Adv. Synth. Catal., 2007, 349, 93 CrossRef CAS.
- (a) G. Gerstberger and R. Anwander, Microporous Mesoporous Mater., 2001, 44, 303 Search PubMed; (b) R. Petroff Saint-Arroman, B. Didillon, A. de Mallmann, J.-M. Basset and F. Lefebvre, Appl. Catal., A, 2008, 337, 78 Search PubMed.
- (a) X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2005, 127, 14015 CrossRef CAS; (b) A. Poater, X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2007, 129, 8207 CrossRef CAS.
- A. M. Leduc, A. Salameh, D. Soulivong, M. Chabanas, J.-M. Basset, C. Copéret, X. Solans-Monfort, E. Clot, O. Eisenstein, V. P. W. Böhm and M. Röper, J. Am. Chem. Soc., 2008, 130, 6288 CrossRef CAS.
- (a) F. Blanc, N. Rendon, R. Berthoud, J.-M. Basset, C. Copéret, Z. J. Tonzetich and R. R. Schrock, Dalton Trans., 2008, 3156 RSC; (b) N. Rendon, R. Berthoud, F. Blanc, D. Gajan, T. Maishal, J.-M. Basset, C. Copéret, A. Lesage, L. Emsley, S. C. Marinescu, R. Singh and R. R. Schrock, Chem.–Eur. J., 2009, 15, 5083 CrossRef CAS.
- (a) S. J. Malcolmson, S. J. Meek, E. S. Sattely, R. R. Schrock and A. H. Hoveyda, Nature, 2008, 456, 933 CrossRef CAS; (b) M. M. Flook, A. J. Jiang, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962 CrossRef CAS.
- (a) C. Copéret, O. Maury, J. Thivolle-Cazat and J.-M. Basset, Angew. Chem., Int. Ed., 2001, 40, 2331 CrossRef CAS; (b) E. Le Roux, M. Chabanas, A. Baudouin, A. de Mallmann, C. Copéret, E. A. Quadrelli, J. Thivolle-Cazat, J.-M. Basset, W. Lukens, A. Lesage, L. Emsley and G. J. Sunley, J. Am. Chem. Soc., 2004, 126, 13391 CrossRef CAS; (c) F. Blanc, C. Copéret, J. Thivolle-Cazat and J.-M. Basset, Angew. Chem., Int. Ed., 2006, 45, 6201 CrossRef CAS; (d) F. Blanc, J. Thivolle-Cazat, J.-M. Basset and C. Copéret, Chem.–Eur. J., 2008, 14, 9030 Search PubMed.
- This reaction can also be performed with the combination of dehydrogenation and alkene metathesis catalysts, see: (a) R. L. Burnett and T. R. Hughes, J. Catal., 1973, 31, 55 CrossRef CAS; (b) A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257 CrossRef CAS.
- (a) V. A. Zakharov, V. K. Dudchenko, E. Paukstis, L. G. Karakchiev and Y. I. Yermakov, J. Mol. Catal., 1977, 2, 421 CrossRef CAS; (b) J. Schwartz and M. D. Ward, J. Mol. Catal., 1980, 8, 465 CrossRef CAS; (c) Y. I. Yermakov, Y. A. Ryndin, O. S. Alekseev, D. I. Kochubei, V. A. Shmachkov and N. I. Gergert, J. Mol. Catal., 1989, 49, 121 Search PubMed; (d) V. Vidal, A. Theolier, J. Thivolle-Cazat, J.-M. Basset and J. Corker, J. Am. Chem. Soc., 1996, 118, 4595 CrossRef CAS; (e) F. Rataboul, A. Baudouin, C. Thieuleux, L. Veyre, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2004, 126, 12541 CrossRef CAS.
- (a) C. Lécuyer, F. Quignard, A. Choplin, D. Olivier and J. M. Basset, Angew. Chem., Int. Ed. Engl., 1991, 30, 1660 CrossRef; (b) M. Chabanas, V. Vidal, C. Copéret, J. Thivolle-Cazat and J.-M. Basset, Angew. Chem., Int. Ed., 2000, 39, 1962 CrossRef CAS.
- V. R. Dufaud and J.-M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806 CrossRef CAS.
- (a) V. Vidal, A. Theolier, J. Thivolle-Cazat and J.-M. Basset, Science, 1997, 276, 99 CrossRef CAS; (b) J.-M. Basset, C. Copéret, D. Soulivong, M. Taoufik and J. Thivolle-Cazat, Acc. Chem. Res., 2010, 43, 323 CrossRef CAS; (c) C. Copéret, Chem. Rev., 2010, 110, 656 CrossRef CAS.
- (a) D. Soulivong, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, B. M. Maunders, R. B. A. Pardy and G. J. Sunley, Angew. Chem., Int. Ed., 2004, 43, 5366 CrossRef CAS; (b) D. Soulivong, S. Norsic, M. Taoufik, C. Copéret, J. Thivolle-Cazat, S. Chakka and J.-M. Basset, J. Am. Chem. Soc., 2008, 130, 5044 CrossRef CAS.
- P. Avenier, M. Taoufik, A. Lesage, X. Solans-Monfort, A. Baudouin, A. de Mallmann, L. Veyre, J.-M. Basset, O. Eisenstein, L. Emsley and E. A. Quadrelli, Science, 2007, 317, 1056 CrossRef CAS.
- P. Euzen, P. Raybaud, X. Krokidis, H. Toulhoat, J.-L. Le Loarer, J.-P. Jolivet and C. Froidefond, in Handbook of Porous Solids, ed. F. Schüth, K. Sing and J. Weitkamp, Wiley-VCH, Weinheim, 2008, 1591–1677 Search PubMed.
- L. K. Hudson, C. Misra, A. J. Perrotta, K. Wefers and F. S. Williams, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000, vol. 2, 339–358 Search PubMed.
- M. Digne, P. Raybaud, P. Sautet, B. Rebours and H. Toulhoat, J. Phys. Chem. B, 2006, 110, 20719 Search PubMed.
- X. Krokidis, P. Raybaud, A. E. Gobichon, B. Rebours, P. Euzen and H. Toulhoat, J. Phys. Chem. B, 2001, 105, 5121 CrossRef CAS.
- (a) M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2002, 211, 1 CrossRef CAS; (b) M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54 CrossRef CAS.
- (a) J. P. Beaufils and Y. Barbaux, J. Chim. Phys. Phys.- Chim. Biol., 1981, 78, 347 CAS; (b) P. Nortier, P. Fourre, A. B. M. Saad, O. Saur and J. C. Lavalley, Appl. Catal., 1990, 61, 141 Search PubMed.
- H. Knözinger and P. Ratnasamy, Catal. Rev. Sci. Eng., 1978, 17, 31 CrossRef.
- R. Wischert, C. Copéret, F. Delbecq and P. Sautet, Angew. Chem., Int. Ed., 2011, 50, 3202 Search PubMed.
- J. Peri, J. Phys. Chem., 1966, 70, 3168 CAS.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J.-M. Basset, J. Phys. Chem. B, 2006, 110, 23944 CrossRef CAS.
- (a) J. G. Larson and W. K. Hall, J. Phys. Chem., 1965, 69, 3080 CAS; (b) P. J. Robertson, M. S. Scurrell and C. Kemball, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 903 RSC; (c) L. Quanzhi and Y. Amenomiya, Appl. Catal., 1986, 23, 173 Search PubMed; (d) J. S. J. Hargreaves, G. J. Hutchings, R. W. Joyner and S. H. Taylor, Appl. Catal., A, 2002, 227, 191 CrossRef CAS.
- A recent investigation has highlighted the role of these Lewis acid sites in the non-dissociative adsorption of CO and H2 on alumina pre-treated at high temperature: E. N. Gribov, O. Zavorotynska, G. Agostini, J. G. Vitillo, G. Ricchiardi, G. Spoto and A. Zecchina, Phys. Chem. Chem. Phys., 2010, 12, 6474 Search PubMed.
- (a) W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 351 CrossRef CAS; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CAS.
- (a) R. L. Burwell, J. Catal., 1984, 86, 301 CrossRef; (b) D. G. H. Ballard, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2191 Search PubMed; (c) P. J. Toscano and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 653 CrossRef CAS.
- J. Joubert, F. Delbecq, P. Sautet, E. Le Roux, M. Taoufik, C. Thieuleux, F. Blanc, C. Copéret, J. Thivolle-Cazat and J.-M. Basset, J. Am. Chem. Soc., 2006, 128, 9157 CrossRef CAS.
- (a) K. H. Dahmen, D. Hedden, R. L. Burwell and T. J. Marks, Langmuir, 1988, 4, 1212 CrossRef CAS; (b) M. Jezequel, V.r. Dufaud, M. J. Ruiz-Garcia, F. Carrillo-Hermosilla, U. Neugebauer, G. P. Niccolai, F. Lefevre, F. Bayard, J. Corker, S. Fiddy, J. Evans, J.-P. Broyer, J. Malinge and J.-M. Basset, J. Am. Chem. Soc., 2001, 123, 3520 CrossRef CAS.
- M. Delgado, C. C. Santini, F. Delbecq, R. Wischert, B. Le Guennic, G. Tosin, R. Spitz, J.-M. Basset and P. Sautet, J. Phys. Chem. C, 2010, 114, 18516 Search PubMed.
- M. S. Eisen and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 10358 CrossRef CAS.
- (a) A. Salameh, J. Joubert, A. Baudouin, W. Lukens, F. Delbecq, P. Sautet, J.-M. Basset and C. Copéret, Angew. Chem., Int. Ed., 2007, 46, 3870 CrossRef CAS; (b) A. Salameh, A. Baudouin, J.-M. Basset and C. Copéret, Angew. Chem., Int. Ed., 2008, 47, 2117 CrossRef CAS; (c) R. Wischert, C. Copéret, F. Delbecq and P. Sautet, ChemCatChem, 2010, 2, 812 Search PubMed.
- (a) J. C. Mol, Catal. Today, 1999, 51, 289 CrossRef CAS; (b) J. C. Mol, Top. Catal., 2004, 27, 97 CrossRef CAS; (c) J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39 CrossRef CAS.
- E. Verkuijlen, F. Kapteijn, J. C. Mol and C. Boelhouwer, J. Chem. Soc., Chem. Commun., 1977, 198 RSC.
- (a) X. Y. Chen, X. Y. Zhang and P. Chen, Angew. Chem., Int. Ed., 2003, 42, 3798 CrossRef CAS; (b) X. Zhang, X. Chen and P. Chen, Organometallics, 2004, 23, 3437 CrossRef CAS; (c) S. Narancic and P. Chen, Organometallics, 2004, 24, 10 Search PubMed; (d) A. Salameh, C. Copéret, J.-M. Basset, V. P. W. Böhm and M. Röper, Adv. Synth. Catal., 2007, 349, 238 CrossRef CAS.
- (a) F. Kapteijn, H. L. G. Bredt, E. Homburg and J. C. Mol, Ind. Eng. Chem. Prod. Res. Dev., 1981, 20, 457 Search PubMed; (b) Y. Chauvin and D. Commereuc, J. Chem. Soc., Chem. Commun., 1992, 462 RSC.
- (a) W. A. Herrmann, W. Wagner, U. N. Flessner, U. Volkhardt and H. Komber, Angew. Chem., Int. Ed. Engl., 1991, 30, 1636 CrossRef; (b) F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149 CrossRef; (c) A. Salameh, A. Baudouin, D. Soulivong, V. Böhm, M. Röper, J.-M. Basset and C. Copéret, J. Catal., 2008, 253, 180 Search PubMed.
- W. A. Herrmann, J. G. Kuchler, J. K. Felixberger, E. Herdtweck and W. Wagner, Angew. Chem., Int. Ed. Engl., 1988, 27, 394 CrossRef.
- (a) E. Le Roux, M. Taoufik, C. Copéret, A. de Mallmann, J. Thivolle-Cazat, J.-M. Basset, B. M. Maunders and G. J. Sunley, Angew. Chem., Int. Ed., 2005, 44, 6755 CrossRef CAS; (b) M. Taoufik, E. Le Roux, J. Thivolle-Cazat, C. Copéret, J.-M. Basset, B. Maunders and G. J. Sunley, Top. Catal., 2006, 40, 65 CrossRef CAS.
- J. Joubert and P. Sautet, unpublished results.
- C. Copéret, Pure Appl. Chem., 2009, 81, 585 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |