The spectroscopy and thermochemistry of phenylallyl radical chromophores†
Tyler P.
Troy
a,
Nahid
Chalyavi
a,
Ambili S.
Menon
abc,
Gerard D.
O'Connor
a,
Burkhard
Fückel
a,
Klaas
Nauta
a,
Leo
Radom
ab and
Timothy W.
Schmidt
*a
aSchool of Chemistry, Building F11 The University of Sydney, NSW 2006, Australia. E-mail: timothy.schmidt@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 2781
bARC Centre of Excellence for Free Radical Chemistry and Biotechnology, Australia
cDepartment of Chemistry, University of Fribourg, CH-1700, Fribourg, Switzerland
First published on 27th June 2011
Abstract
The resonant two-color two-photon ionization and laser induced fluorescence excitation spectra of the 1-phenylallyl (cinnamyl) and inden-2-ylmethyl radicals are reported. The 1-phenylallyl radical is found to fluoresce with low yield, permitting only a coarse dispersed fluorescence spectrum, while the inden-2-ylmethyl radical yields sufficient fluorescence to obtain ground-state vibrational frequencies and two-dimensional fluorescence spectra. Computed ionization energies and thermochemical properties including radical stabilization energies are reported for a range of resonance-stabilized radicals, including the phenylpropargyl, vinylpropargyl and phenylallyl radicals.
Introduction
Complex and energetic environments such as plasmas, flames, planetary atmospheres and interstellar space contain exotic chemical species such as hydrocarbon radicals and ions.1–3 Among the radicals most resistant to passivation by hydrogen addition are the resonance-stabilized radicals (RSRs). These species may be depicted by several Lewis structures that contribute to the description of the electronic structure. Well-known examples include the allyl (˙CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), propargyl (˙CH2C
CH2), propargyl (˙CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) and benzyl (˙CH2Ph) radicals (Fig. 1). Such species have been demonstrated to be participants in the chemistry of fuel-rich combustion, with propargyl radical widely believed to build up to high concentrations in such environments, eventually reacting with another propargyl radical to produce benzene and subsequently soot.4,5
CH) and benzyl (˙CH2Ph) radicals (Fig. 1). Such species have been demonstrated to be participants in the chemistry of fuel-rich combustion, with propargyl radical widely believed to build up to high concentrations in such environments, eventually reacting with another propargyl radical to produce benzene and subsequently soot.4,5
 | ||
| Fig. 1 Resonance-stabilized radical structures. | ||
A commonly-used measure of the chemical stability of a hydrocarbon radical of this type (˙CH2X) is the radical stabilization energy (RSE),6–8 which is often defined as the energy change in the hydrogen-atom-transfer reaction:
| ˙CH2X + CH4 → CH3X + ˙CH3 |
| RSE[R˙] = BDE[CH3 − H] − BDE[XCH2 − H] |
Methyl radicals disubstituted by such groups are found to be stabilized to an even greater extent, though the effect is generally less than additive.8 It is convenient in this respect to define the deviation from additivity of the RSEs (DARSE) for a disubstituted radical (˙CHXY) by the equation:
| DARSE(˙CHXY) = RSE(˙CHXY) − RSE(˙CH2X) − RSE(˙CH2Y) |
In 2008 we reported the discovery of the 1-phenylpropargyl radical (Fig. 1, 1PPR) as a product of an electrical discharge through benzene seeded in argon.11,12 This radical was found to be the most conspicuous fluorescent species in this environment after the ever-present C2, C3 and CH molecules. In addition, another product of this and other hydrocarbon discharges was identified as the cis- and trans-1-vinylpropargyl radicals13 (Fig. 1, 1VPR). These species may be viewed, respectively, as disubstituted methyl radicals with phenyl and ethynyl substituents (1PPR) or vinyl and ethynyl substituents (1VPR). They could alternatively be named α-ethynylbenzyl radical (1PPR) and ethynylallyl radical (1VPR). As we shall report, the combined effect of the two substituents leads to substantial radical stabilization energies (approximately 100 kJ mol−1) for both 1PPR and 1VPR.
The identification of 1PPR and 1VPR as abundant discharge products suggests that a Ph˙CHCHCH2 species, 1-phenylallyl or cinnamyl (the name by which it is commonly referred, Fig. 1, 1PAR), should be formed from a discharge containing a suitable precursor. Such a species could be considered a disubstituted methyl radical with phenyl and vinyl substituents and would complete this family of RSR systems. It could be considered to be partly allylic, and partly benzylic. In this paper, we report the identification of the cinnamyl radical (t-1PAR) in the products of an electrical discharge through argon containing traces of trans-β-methylstyrene. We also identify a related chromophore derived from 2-methylindene, the inden-2-ylmethyl radical (Fig. 1, I2MR). In a similar manner to the 1PPR and 1VPR radicals, these species are identified through the synergistic application of several spectroscopic techniques: resonant two-color two-photon ionization, laser induced fluorescence, dispersed fluorescence and two-dimensional fluorescence spectroscopy, as well as the application of several quantum chemical methods. Calculated radical stabilization energies and related thermochemical quantities are presented for several of these radicals. A positive identification of the trans-1-phenylallyl radical (t-1PAR) and the related chromophore, the benzylallenyl radical, is given in the accompanying paper by Sebree et al.36
Experimental techniques
A pulsed discharge nozzle was used to generate the radical of interest from a relevant precursor. A more thorough description of a pulsed discharge nozzle is provided by Oshima and Endo.14 For the trans-1-phenylallyl radical, the vapour pressure of trans-β-methylstyrene (Aldrich, 99%) was seeded into 10 bar of argon at 298 K. For the inden-2-ylmethyl radical, 2-methylindene (Aldrich, 98%) was heated to 313 K and seeded into 5 bar of argon. The seeded beam was supersonically expanded into a vacuum and passed between two high-voltage electrodes. A −1.5 to −2.0 kV pulse of 40 to 120 μs duration was applied to the inner electrode of the pulsed discharge nozzle through 15 to 40 kΩ of resistance and timed to coincide with the gas pulse. For the fluorescence experiments, the nascent radicals present in the molecular beam were probed ∼2 cm downstream of the pulsed discharge nozzle with the output from a Nd:YAG-pumped dye laser circulating Coumarin 503. The laser induced fluorescence was imaged onto the entrance slit of a 0.75 m monochromator operated with a slit width of 3 mm. Fluorescence was detected by a photomultiplier tube. A fluorescence lifetime measurement for I2MR was recorded over 2000 laser shots. Dispersed fluorescence spectra were measured by fixing the pump laser frequency to a transition of the radical of interest while scanning the monochromator. The monochromator was calibrated between 14000 and 22000 cm−1 with a sodium lamp, using known emission lines of sodium and argon. The fluorescence signal was digitized with a 500 MHz oscilloscope and transferred to a personal computer to be processed using in-house software. Relative instrument timings were controlled by a digital delay generator operated at a repetition rate of 20 Hz.Two-dimensional (2D) fluorescence spectra were recorded using a Princeton Instruments PI-MAX spectrometer. The fluorescence emission was dispersed in wavelength across a CCD array exposed for 20 laser shots, producing a 1D spectrum of intensity vs. emission wavelength over the wavelength range 464–734 nm (∼21550–13620 cm−1). A 2D map of emission vs. excitation was produced by stepping the laser between 517–480 nm (∼19340–20830 cm−1).
The resonant two-color two-photon ionization spectra were measured in a two-stage differentially-pumped vacuum chamber. A supersonically expanded beam, produced by a pulsed discharge nozzle (referenced above) was skimmed and exposed to counter-propagating laser beams passing between the grids of a time-of-flight mass spectrometer. Excitation photons were provided from the output of a XeCl excimer laser pumped dye laser circulating Coumarin 503. Ionizing radiation was provided from the output of a GAM ArF excimer laser (193 nm). The positive ions, accelerated by 2.4 kV, were detected by a tandem micro-channel plate. Ion signals from m/z 117 and m/z 129 were recorded as a function of excitation wavelength. Absolute wavelength calibration of the tunable laser was obtained using a Coherent Wavemaster wavemeter. Photoionization efficiency (PIE) curves were obtained by scanning the frequency-doubled output of a Nd:YAG-pumped dye laser as the second, ionizing photon after origin (000) excitation. Therefore the PIE curves represent an isomer-selected, 0 K ensemble. A lifetime measurement for t-1PAR was recorded by scanning the excitation laser in time relative to the ionization laser.
Computational methods
Spectroscopy
Density functional theory (DFT) calculations using the B3-LYP functional were performed with the Gaussian 03 package.15 Optimized geometries and normal mode frequencies were obtained using the 6-311++G(d,p) basis set. Excited-states were calculated ab initio with the GAMESS quantum chemistry package16 at the complete-active-space self-consistent-field (CASSCF) level of theory, with multi-reference second-order perturbation theory (PT2) used to account for dynamic correlation (CASSCF+PT2). The active spaces of the radical systems with Cs symmetry were chosen to contain only a′′ orbitals so that all excitations within the active space gave rise to configurations of A′′ symmetry. The 6-311G(d,p) basis set was used in these calculations.Thermochemistry
Calculations were carried out with the Gaussian 03,15 Gaussian 0917 and Molpro 2008.118 packages. Geometries of radicals (˙CHXY) and their molecular precursors (CH2XY) were optimized at the B3-LYP/6-31G(2df,p) level. Single-point energies at 0 K were then obtained with the high-level G3X(MP2)-RAD composite procedure,19 which approximates the URCCSD(T)/G3XLarge level of theory. This has been found to produce reliable radical stabilization energies (RSEs) for carbon-centered radicals.8,10Ionization energies
Vertical ionization energies (IEs) were calculated using B3-LYP with a range of basis sets. As the basis set increases in size, we find that the calculated B3-LYP IEs increase and appear to reach a converged value for B3-LYP well before our ultimate 6-311++G(3df,3pd) basis. The B3-LYP/6-311++G(3df,3pd) procedure, as well as G3X(MP2)-RAD, were then applied generally to calculate the IEs for a range of RSRs. In these calculations, the zero-point vibrational energy in the vertically ionized cationic state was assumed to be the same as that in the radical ground state, with the result that it cancels from the ionization energy determination.Results and discussion
Identification of the inden-2-ylmethyl radical
Fig. 2 shows the excitation spectrum observed from the discharge products of 2-methylindene in the wavelength range 520–485 nm (∼19230–20620 cm−1). The resonant two-color two-photon ionization spectrum was recorded while monitoring m/z 129 and shows a dominant origin-band at 19365 cm−1. If we assume that the connectivity of the carbon skeleton is preserved, this mass may be accounted for by two resonance-stabilized radical isomers, inden-2-ylmethyl radical (I2MR) and 2-methylindenyl radical (2MIR) (see Fig. 1). The Zwier group recently observed an analogous chromophore to I2MR, the 1-hydronaphthyl radical, which exhibits a dominant origin-band at 18949 cm−1, within 2% of the energy of the origin observed for m/z 129.20 For the indenyl chromophore, exhibited by 2MIR (Fig. 1), the only relevant previous spectrum was recorded by Inoue et al.21 It shows excitation and fluorescence centered at 415 nm (∼24100 cm−1). As such, the weight of experimental evidence is in favor of I2MR as the carrier in the present experiments. In order to assist with the differentiation, we carried out ab initio calculations of the vertical excitation energy. As shown in Table 1, multi-reference perturbation theory predicts the vertical transition of I2MR to be in the 20000 cm−1 range, consistent with the expectations of a vinyl-substituted benzyl radical. The benzyl radical absorbs near 22000 cm−1,22 and vinyl substitution normally red-shifts the absorption position of conjugated π-chromophores by about 2000 cm−1. Contrastingly, the 2MIR radical has an altogether different electronic structure. The cyclopentadienyl radical has a degenerate ground state, and its derivatives thus exhibit low-lying excited-states. CASSCF+PT2 calculations with a [5,5] active space were performed on 2MIR, with the two lowest states weighted equally to achieve convergence. The two lowest-energy excitations were found near 11000 and 16000 cm−1. This is not consistent with either the present observation of a sharp band system near 19000 cm−1 or with the previous observations of Inoue et al.21 Theoretical considerations of the excitation energy thus also favor the I2MR isomer. The predicted vertical excitation is about 1000 cm−1 higher than the observed origin. To compare directly with the experimental origin position, one should also account for geometrical relaxation and zero-point energy reduction in the excited-state, both of which would have the effect of lowering our calculated excitation energies. However, as our goal here is to guide the spectral assignment, we are satisfied that our approximate calculated excitation energies can exclude the 2MIR isomer, and support I2MR as the spectral carrier.| Method | I2MR | trans-1PAR | cis-1PAR |
|---|---|---|---|
| [3,3] | 21871 | 22202 | 26517 |
| [5,5] | 21574 | 21497 | 25093 |
| [7,7] | 22421 | 22197 | 21573 |
| [9,9] | 21494 | 21327 | 21529 |
| [3,3]PT2 | 23767 | 25295 | 22045 |
| [5,5]PT2 | 18603 | 18616 | 23050 |
| [7,7]PT2 | 20466 | 20586 | 20210 |
| [9,9]PT2 | 20257 | 20325 | 20349 |
| Exp. | 19365 | 19208 | — |
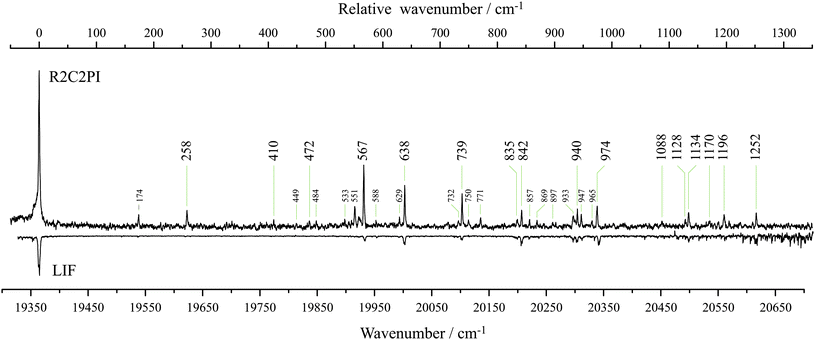 | ||
| Fig. 2 The resonant two-color two-photon ionization (R2C2PI) excitation spectrum recorded while monitoring m/z 129 here reported to be carried by the inden-2-ylmethyl radical (I2MR). The same excitation spectrum is observed by laser induced fluorescence (LIF). Large labels denote bands assigned to single quanta transitions of a′ modes. Small labels indicate combination bands. Assignments are given in Table 2. | ||
In our previous work with vinylpropargyl, phenylpropargyl and 1-indanyl, the dispersed fluorescence spectra, in conjunction with DFT frequency calculations, were found to be of great assistance in arriving at a conclusive chemical assignment.12,13,28Fig. 3 shows the spectrum of dispersed fluorescence resulting from origin-band excitation. The fluorescence was observed to exhibit a lifetime of 120 ns. Comparison of the observed frequencies with those calculated for I2MR and 2MIR (see ESI†) reveals that only I2MR can account for all of the observed frequencies. As has been shown to be effective in the past,12,13 ground-state frequencies were scaled by 0.97. We find that the scaled calculated I2MR frequencies show good agreement with experiment, allowing for facile assignment of the origin dispersed fluorescence spectrum. These assignments are given in Table 2. All of the most intense features (large labels, Fig. 3) are assigned as transitions to single quanta of a′ modes. The weakest assigned a′ mode, ν14, barely discernable from noise at 1409 cm−1, represents the symmetric bending mode of the out-of-plane hydrogens. It is not expected to couple strongly to the π* ← π electronic transition. Modes of a′ symmetry from ν8 to ν1 represent C–H stretching modes which also will not couple strongly to the electronic transition. This is observed experimentally as evidenced in the inset of Fig. 3 which shows emission recorded up to 3500 cm−1. Here we see an abrupt end to any strong emission beyond ν9 at 1580 cm−1 and no evidence of the a′ C–H stretching modes. Some of the weaker bands assigned to single quanta of a′ modes might also be plausibly assigned to transitions involving multiple quanta. For example, the band at 1292 cm−1 assigned to a single quantum of ν18 could be accounted for by two quanta of ν30. As such, many of the weaker band assignments are considered tentative. Similarly, the multitude of very weak bands that are observed beyond ν9 are likely to be due to combination bands. Since they appear so weakly, and are likely to be plagued by anharmonic coupling, no attempt was made to assign them. The less intense features observed below ν9 not attributable to single quanta of a′ modes, can be assigned to transitions involving multiple quanta of a′′ modes, as given in Table 2. These combination bands are indicated with small peak labels in Fig. 3. The peaks at 205, 528, 815 and 895 cm−1, can be assigned to two quanta of modes ν51, ν49, ν48 and ν47, respectively. The expanded region shows a group of congested peaks for which the assignments are least certain. The two peaks at 1128 and 1140 cm−1 have been assigned to single quanta of modes ν23 and ν22 due to their intensity. The shoulders have been assigned to multiple quanta of a′′ modes which likely borrow intensity from the close-lying a′ modes in Fermi resonance. Combination bands are assigned purely on their similarity to the calculated frequencies and should therefore be considered tentative.
| D0 | D1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp. | Calc. | Assign. | Exp. | Calc. | Assign. | Exp. | Calc. | Assign. | Exp. | Calc. | Assign. |
| a Scaling factors of 0.97 (D0) and 0.95 (D1), see text. | |||||||||||
| 263 | 260 | 3401 | 1377 | 1367 | [1501] | 258 | 255 | 3410 | 629 | 627 | [47105010] |
| 437 | 436 | 3301 | 1409 | 1408 | [1401] | 410 | 427 | 3310 | 732 | 718 | [34103210] |
| 476 | 473 | 3201 | 1432 | 1431 | 1301 | 472 | 463 | 3210 | 857 | 870 | [44105010] |
| 584 | 582 | 3101 | 1463 | 1455 | [1201] | 567 | 570 | 3110 | 869 | 880 | [4720] |
| 650 | 647 | 3001 | 1481 | 1489 | 1101 | 638 | 634 | 3010 | 897 | 889 | [34103010] |
| 787 | 781 | 2901 | 1559 | 1554 | [1001] | 739 | 765 | 2910 | 933 | 939 | [42105010] |
| 848 | 847 | 2801 | 1580 | 1572 | 901 | 835 | 830 | 2810 | 947 | 963 | [41105010] |
| 861 | 862 | 2701 | 205 | 206 | 5102 | 842 | 844 | 2710 | |||
| 955 | 951 | 2601 | 528 | 530 | 4902 | 940 | 932 | 2610 | |||
| 1008 | 1005 | 2501 | 602 | 600 | 48015001 | 974 | 984 | 2510 | |||
| 1095 | 1084 | 2401 | 815 | 818 | 4802 | 1088 | 1061 | 2410 | |||
| 1128 | 1134 | 2301 | 895 | 898 | [4702] | 1128 | 1130 | 2210 | |||
| 1140 | 1153 | 2201 | 962 | 959 | [42015001] | 1134 | 1135 | 2110 | |||
| 1168 | 1159 | 2101 | 993 | 983 | [41015001] | 1170 | 1162 | 2010 | |||
| 1190 | 1186 | 2001 | 1126 | 1126 | 30013201 | 1196 | 1213 | 1910 | |||
| 1242 | 1238 | 1901 | 1136 | 1124 | [38015001] | 1252 | 1258 | 1810 | |||
| 1292 | 1285 | [1801] | 1143 | 1142 | [37015001] | 174 | 202 | 5120 | |||
| 1312 | 1304 | [1701] | 1220 | 1223 | [44014601] | 533 | 516 | 4920 | |||
| 1341 | 1341 | 1601 | 1263 | 1263 | 29013201 | 588 | 588 | [48015010] | |||
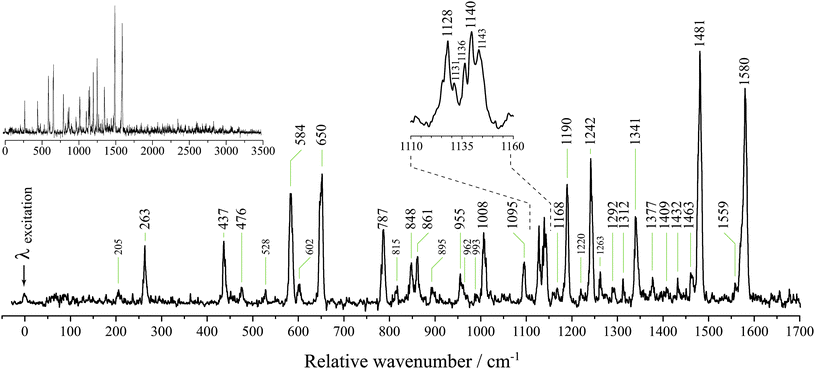 | ||
| Fig. 3 The dispersed fluorescence resulting from origin-band excitation of m/z 129 here reported to be carried by the inden-2-ylmethyl radical (I2MR). Large peak labels indicate bands assigned to emission to single quanta of a′ modes and small labels indicate bands assigned to emission to multiple quanta. The top-left inset shows emission out to 3500 cm−1. Due to the overpowering signal from laser scatter at the excitation wavelength, the detector was suppressed while the monochromator was scanned over this wavelength. As a result, the intensity of the dominant emission at the origin wavelength cannot be discerned. The 2D fluorescence spectrum (Fig. 5) better reflects the true intensity ratios. | ||
The assignment of I2MR as the carrier is further evidenced by the observed photoionization efficiency (PIE) curve presented in Fig. 4, which shows the ionization energy (IE) of the carrier to be 6.737 eV. The rapid onset of ionization indicates little difference between adiabatic and vertical IE. Vertical IEs were calculated using B3-LYP with a range of basis sets, with the results plotted in Fig. 4. As the basis set increases in size, we find that the calculated B3-LYP IEs increase and appear to reach a converged value for B3-LYP well before our ultimate 6-311++G(3df,3pd) basis. The B3-LYP/6-311++G(3df,3pd) procedure, as well as the high-level G3X(MP2)-RAD procedure, were then applied to calculate the IEs for a range of RSRs, with the results displayed in Table 3. We find that B3-LYP/6-311++G(3df,3pd) slightly underestimates the IEs of RSRs, with a mean deviation (MD) from experimental values of −0.08 eV and a mean absolute deviation (MAD) of 0.08 eV. On the other hand, G3X(MP2)-RAD is found to slightly overestimate the IEs, with an MD of +0.07 eV and a MAD of 0.09 eV. This is very helpful in making structural assignments because the two theoretical values generally bracket the experimental value. In the case of the indenylmethyl systems, comparison of the observed vertical IE of 6.737 eV with the calculated B3-LYP/6-311++G(3df,3pd) and G3X(MP2)-RAD values for I2MR (6.67 and 6.84 eV, respectively) and 2MIR (7.39 and 7.50 eV, respectively) thus strongly favor the I2MR isomer. We note in addition that our theoretical results for the 1-phenylpropargyl radical suggest that the current experimental estimate of 7.4 eV is probably too high and deserves re-examination. Indeed, in our previous experiments we ionized 1PPR with a combined photon energy of 7.27 eV, which is within the range suggested by the calculations and can be considered an upper limit.
| Radical | Label | Calcb | Calcc | Expt | Calcb – Expt | Calcc – Expt |
|---|---|---|---|---|---|---|
| a Vibrationless values. b B3-LYP/6-311++G(3df,3pd). c G3X(MP2)-RAD. d This work. e Mean absolute deviation (MAD), mean deviation (MD), largest deviation (LD) from experimental values. | ||||||
| Propargyl | 8.70 | 8.71 | 8.71(2)23 | −0.01(2) | +0.00(2) | |
| Allyl | 8.12 | 8.21 | 8.13146(25)24 | −0.01 | +0.08 | |
| trans-Vinylpropargyl | t-VPR | 7.71 | 7.86 | 7.83(5) 25 | −0.12(5) | +0.03(5) |
| Benzyl | 7.21 | 7.32 | 7.2487(6)26 | −0.04 | +0.07 | |
| 1-Phenylpropargyl | 1PPR | 7.13 | 7.28 | 7.4(1)27 | −0.27(10) | −0.12(10) |
| 3-Phenylpropargyl | 7.14 | 7.29 | 7.2(5)27 | −0.06(50) | +0.09(50) | |
| 1-Indanyl | 6.53 | 6.69 | 6.578(1)28 | −0.05 | +0.11 | |
| trans-1-Phenylallyl | t-1PAR | 6.81 | 6.99 | 6.906(1)d | −0.10 | +0.08 |
| Inden-2-ylmethyl | I2MR | 6.67 | 6.84 | 6.737(1)d | −0.07 | +0.10 |
| 1-Hydronaphthyl | 6.48 | 6.68 | 6.570(1)20 | −0.09 | +0.11 | |
| 2-Hydronaphthyl | 6.41 | 6.60 | 6.487(1)20 | −0.07 | +0.11 | |
| 1,2,3-Trihydronaphthyl | 6.56 | 6.73 | 6.620(1)20 | −0.06 | +0.11 | |
| MAD e | 0.08 | 0.07 | ||||
| MDe | −0.08 | +0.09 | ||||
| LD e | −0.27(10) | −0.12(10) | ||||
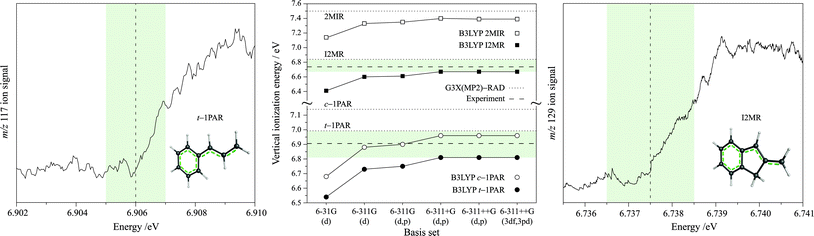 | ||
| Fig. 4 The left and right panels present the photoionization efficiency (PIE) curves recorded while monitoring mass channels m/z 117 (trans-1-phenylallyl radical, t-1PAR) and m/z 129 (inden-2-ylmethyl radical, I2MR), respectively. A conservative confidence interval of 1 meV is shaded. The center panel demonstrates the effect of the basis set on the B3-LYP calculated vertical ionization energies of the candidate carriers. The horizontal dotted lines show the IEs calculated with the G3X(MP2)-RAD procedure. The horizontal dashed lines indicate the experimentally-determined IEs of the inden-2-ylmethyl radical (top) and the trans-1-phenylallyl radical (bottom). We find that the two levels of theory bracket the experimental IE of both systems as is indicated by the shaded region. | ||
Excited-state assignments are less easy to make because of the difficulty in calculating accurate excited-state frequencies. In order to unequivocally make assignments, it is necessary to disperse the fluorescence from each excitation band in order to determine how a particular vibrational wave function maps onto the ground-state vibrational modes. For origin-dominated transitions, the mapping of this coupling is less complicated because there is little change in the geometry of the system upon excitation and normally little change in the excited-state vibrational modes. In this case, the wave functions of the two states involved in a fluorescent transition show the greatest overlap between states with the same number of quanta in similar vibrational modes. The D1 ← D0 transition of I2MR is a good example of an origin-dominated excitation showing a high degree of verticality, as evidenced by the 2D fluorescence spectrum presented in Fig. 5. This spectrum shows both the emission and excitation energies of I2MR relative to its origin-band. The inset spectra opposing either axis show excitation and emission spectra recorded by standard laser induced fluorescence and dispersed fluorescence. We first note the absence of the C2 and CH emissions that are commonly observed in discharge environments. The observed features can be confidently assigned to a single carrier. For almost all of the observed vibronic excitation energies, we see emission back to the same energy in the ground state, indicating that both the excitation and emission spectra will show similar frequencies for that mode. These vertical transitions are emphasized by the diagonal stripe superimposed upon the spectrum. We also notice that not all of the modes are so well behaved. For example, the transition between 700 and 800 cm−1 does not emit to the same energy at which it was excited. By following the dashed vertical and horizontal lines from that transition back to the inset emission and excitation spectra, we can see how the frequency of that particular vibration changes upon excitation. Inspection of the high-resolution excitation and emission spectra (Fig. 2 and Fig. 3, respectively) reveals this to be the 787 cm−1 emission feature and the 739 cm−1 excitation feature, a change of 48 cm−1 upon excitation. In this manner we can map the most prominent bands in the excitation spectrum to their respective ground-state modes, providing assignments for those excited-state frequencies. In order to assign the vibronic structure beyond that observed in 2D fluorescence, we introduce a factor to scale the calculated ground-state frequencies to excited-state frequencies. This factor of 0.98 was derived by minimizing the sum of the squares of differences between the prominent D0 and D1 frequencies from the 2D fluorescence spectrum. By combining this factor with the ground-state scaling factor of 0.97, we have estimated the excited-state frequencies by scaling the calculated harmonic ground-state frequencies by 0.95. This allows us to make the excited-state assignments presented in Table 2.
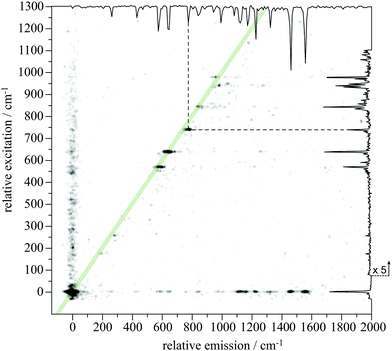 | ||
| Fig. 5 A 2D fluorescence spectrum observed from the discharge of 2-methylindene here reported to be carried by the inden-2-ylmethyl radical (I2MR). Both emission and excitation are plotted relative to the origin. The top and right borders show the emission and excitation spectra recorded by dispersed fluorescence and laser induced fluorescence respectively. The dashed lines indicate how the 787 cm−1 emission feature changes frequency in the excited-state. | ||
Identification of the trans-1-phenylallyl radical
Fig. 6 shows the excitation spectra obtained from an electrical discharge containing trans-β-methylstyrene. The resonant two-color two-photon ionization spectrum was recorded by monitoring m/z 117, consistent with the loss of one hydrogen atom from the precursor. It is identical to that reported by Sebree et al. in the accompanying article.36 As for other aromatic RSRs, the excitation spectra are origin dominated. This is less evident in the resonant two-color two-photon ionization spectrum (R2C2PI), which exhibits signs of saturation. The Franck–Condon activity is most prominently displayed by features 156, 338, 401 and 598 cm−1 from the origin. The laser induced fluorescence spectrum (LIF) is identical to the resonant two-color two-photon ionization spectrum, but was recorded at a lower signal-to-noise ratio. Assignment of these features first requires identification of the carrier of the spectrum. The origin-band at 19208 cm−1 is consistent with the same phenylallyl chromophore exhibited by I2MR and the 1-hydronaphthyl radical. The calculated vertical excitation energies in Table 1 are higher than the observed origin-band by about 1000 cm−1. The discrepancy with the observed excitation energy is very similar to that observed for I2MR. While we expect the trans-β-methylstyrene precursor to yield the trans-1-phenylallyl radical, it is possible that rearrangement may occur in the energetic discharge environment to form either or both stereoisomers about the allylic moiety. The 2-phenylallyl radical shown in Table 5 is ruled out as a potential carrier principally because of the different connectivity of the carbon skeleton. Furthermore, the radical is not delocalized across the phenyl π system and, as is shown later, its RSE is less than that for the parent allyl radical. In our previous work with cis- and trans-1VPR,12 the dispersed fluorescence spectra, in conjunction with DFT frequency calculations, proved invaluable in arriving at a conclusive chemical assignment where “fingerprint” vibrational modes were found to be unique to each particular stereoisomer. In the present study, the difficulty with which the laser induced fluorescence was obtained conferred still greater difficulty in obtaining dispersed fluorescence spectra. The excited-state was observed to have a lifetime of 63 ns, as determined by the method explained in the Experimental methods section. A low-resolution origin dispersed fluorescence (DF) spectrum is presented in the inset of Fig. 6. As the spectrum was recorded with 3 mm slits, it is broad and noisy, and the indicated peak positions are taken as indicative only. It nevertheless demonstrates two prominent features centered at 390 and 1570 cm−1 on either side of several weaker features. Features lower than 250 cm−1 could not be observed due to overpowering laser scatter.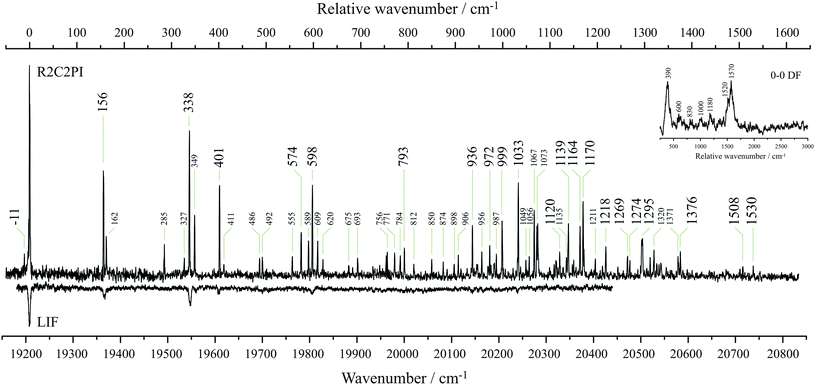 | ||
| Fig. 6 The resonant two-color two-photon ionization excitation spectrum (R2C2PI) was recorded while monitoring m/z 117. It is here reported to be carried by the trans-1-phenylallyl radical. The laser induced fluorescence (LIF) excitation spectrum of the same species recorded with low signal to noise. The top-right inset spectrum shows the dispersed fluorescence (DF) resulting from origin-band excitation. Large labels denote bands assigned to single quanta transitions of a′ modes. Small labels indicate combination bands or unassigned bands. | ||
In order to arrive at a chemical assignment, we turn to the observed ionization energy. We calculate the IEs for trans-1-phenylallyl to be 6.81 eV (B3-LYP/6-311++G(3df,3pd)) and 6.99 eV (G3X(MP2)-RAD). These nicely bracket the experimental value of 6.906 eV derived from the photoionization efficiency trace for the m/z 117 species in Fig. 4. This agrees closely with the work of Zwier's group to be found in the accompanying article.36 For the cis isomer, the calculated IEs are 6.97 eV (B3-LYP) and 7.14 eV (G3X(MP2)-RAD). On the basis of the general behavior of B3-LYP/6-311++G(3df,3pd) and G3X(MP2)-RAD for calculating IEs, the measured IE is in favor of assignment of the trans isomer as the spectral carrier.
Fig. 7 presents the results from a series of B3-LYP/6-311++G(d,p) optimizations of 1-phenylallyl with a fixed dihedral torsional angle between the phenyl and allylic moieties. The torsional angle was stepped from 0 to 180° in 10° increments. This treatment reveals the trans isomer to be stabilized by 1351 cm−1 compared with the cis isomer. Moreover, the barrier to cis–transisomerization exceeds 4000 cm−1. The non-planar minimum for the cis isomer reveals that steric hindrance is likely to be responsible for the higher calculated energy. The trans geometry of the molecular precursor, the observed IE, and the lower energy of the trans isomer of the radical all argue for assignment of the observed spectrum to the trans isomer.
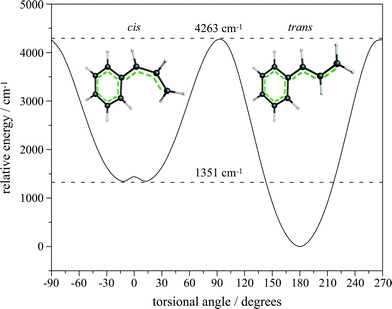 | ||
| Fig. 7 Relaxed scan of the allyl torsion of 1-phenylallyl radical calculated with B3-LYP/6-311++G(d,p). The trans isomer is stabilized by 1351 cm−1 compared with the cis isomer, and the barrier to isomerization is 4263 cm−1. The optimized geometry of each isomer is illustrated. | ||
An attempt may be made to assign the m/z 117 excitation spectrum by scaling calculated ground-state modes by the factor of 0.95 determined for I2MR. We take the lowest five observed bands as a fingerprint of the spectral carrier. The scaled a′ frequencies of trans-1-phenylallyl agree with the observed band position with a mean squared deviation of 10 cm−1, the same as observed for I2MR, the identity of which is certain. Contrastingly, the mean squared deviation in these five lowest bands is twice as large for the cis isomer. Moreover, the diphenylmethyl radical identified by Tsuge and co-workers, which is similarly sterically hindered as is the present cis isomer, demonstrates progressions in torsional modes.29 There is no evidence of a progression in the 60 cm−1 torsional mode that would be characteristic of the cis isomer in the present excitation spectrum, and thus we finally conclude that the carrier is indeed the trans-1-phenylallyl radical. Assignments for the D1 ← D0excitation spectrum are given in Table 4.
| D1 | |||||
|---|---|---|---|---|---|
| Exp. | Calc. | Assign. | Exp. | Calc. | Assign. |
| a ν 48 in D0 state is 92 cm−1. | |||||
| −11a | −2 | 4811 | 1139 | 1129 | [2210] |
| 0 | 0 | 000 | 1164 | 1149 | [2110] |
| 156 | 152 | 3310 | 1170 | 1162 | [2010] |
| 338 | 343 | 3210 | 1218 | 1238 | [1910] |
| 401 | 391 | 3110 | 1269 | 1254 | [1810] |
| 574 | 593 | 3010 | 1274 | 1286 | [1710] |
| 598 | 599 | 2910 | 1295 | 1307 | [1610] |
| 793 | 793 | 2810 | 1376 | 1389 | [1510] |
| 936 | 941 | 2710 | 162 | 173 | 4820 |
| 972 | 958 | [2610] | 285 | 260 | 4720 |
| 999 | 993 | [2510] | 327 | 340 | [48113210] |
| 1033 | 1045 | [2410] | 349 | 328 | 48104610 |
| 1120 | 1119 | [2310] | 411 | 485 | [4620] |
The singly-occupied non-bonding molecular orbitals (SOMOs) of both I2MR and t-1PAR are predominantly housed on the allyl group, with density on the α and γ, and 2, 4 and 6 carbon atoms. The highest doubly-occupied molecular orbitals (HOMOs) are bonding between β and γ, 1 and 2, and, 4 and 5. The LUMOs are antibonding for these same pairs of atoms. The electronic excitation represents the combination of both the HOMO–SOMO and SOMO–LUMO electronic transitions. Both transitions thus reduce the bond order between the aforementioned pairs of atoms. This is borne out by the excited-state geometry calculations by Sebree et al., in the accompanying article.36 All of the strongest bands in the excitation spectrum of t-1PAR can be accounted for by single quanta of a′ bands in the excited-state. The lowest frequency modes observed correspond to changes in bond angles. The band at 156 cm−1 corresponds to in-plane rocking, while the 338 cm−1 excitation may be described as a “concertina” motion of the allyl moiety. The next band at 401 cm−1 also mostly involves this part of the molecule, while the 598 cm−1 feature is due to a distortion of the aromatic ring. No particularly strong features are seen in the 1500 cm−1 region, yet the coarse emission spectrum shows activity at 1570 cm−1 corresponding to CC stretching motion. This same frequency is seen in the phenylallyl species formed upon dissociative adsorption of allylbenzene onto ZnO.30 Yet, there is little evidence for its counterpart in the excitation spectrum, possibly due to anharmonic coupling associated with surmounting of the barrier to trans–cisisomerization in the excited-state.
The are a myriad of combinations of a′′ modes that may plausibly explain many of the weaker features observed in the excitation spectrum. Because there are fewer possibilities at low frequency, we may assign these features with some confidence. The lowest out-of-plane mode, ν48, is calculated at 87 cm−1 and corresponds to a bowing across the long axis of the molecule. In the excited-state, we located a feature at 162 cm−1, which we attribute to two quanta of this vibration, its frequency thus determined to be 81 cm−1. Fermi resonance possibly plays a role in its intensity, being only 6 cm−1 from the 3310 band. The 285 cm−1 feature is assigned to two quanta of the allyl torsional mode, ν47. The calculated frequency of this mode is 130 cm−1, which would predict a band at 260 cm−1. However, in the parent species, trans-β-methylstyrene, this mode increases from just 28 cm−1 to 165 cm−1 in the first excited-state.31 As such, it is not surprising to see a frequency increase for the present species, albeit more modest. The feature at 327 cm−1 is 11 cm−1 below 338 cm−1 (3210), and is taken as a hot band, probably 48113210. Mode ν48 is determined to have a frequency of 81 cm−1 in the excited-state and thus is found to be 92 cm−1 in the ground state. The scaled DFT result is 87 cm−1. The expected 4811 band is indeed identified 11 cm−1 below the origin. The band at 349 cm−1 is stronger than expected, perhaps due to Fermi resonance with the 3210 band. If ν46 exhibits an excited-state frequency of 206 cm−1, then this band would be assigned as 48104610 with 4820 found at 411 cm−1. Other tentatively assigned weaker features are given in Table 4.
Thermochemical considerations
Relative energies at 0 K for radicals (ΔEr) and their parent precursor molecules (ΔEm), and associated bond dissociation energies (BDEs), radical stabilization energies (RSEs), and deviations from additivity of RSEs (DARSEs), calculated at the G3X(MP2)-RAD level, are presented in Table 5. This level of theory has been used successfully previously to study radical thermochemistry.8,19 For example, it produces RSEs for the allyl, propargyl and benzyl radicals of 71.1, 52.9 and 59.2 kJ mol−1, respectively, which compare well with experimental values9 of 69.0 ± 3, 55.0 ± 4.2 and 61.7 ± 5.0 kJ mol−1. There are two possible isomers of the phenylpropargyl radical. 1-phenylpropyne is the precursor of 3-phenylpropargyl radical (3PPR), while 3-phenylpropyne yields 1-phenylpropargyl radical (1PPR). We can see from Table 5 that the RSE of the 1-phenylpropargyl radical (100.8 kJ mol−1) is substantially greater than that for 3-phenylpropargyl radical (62.1 kJ mol−1). This can partly be attributed to the greater stability of 1PPR relative to 3PPR (by 9.5 kJ mol−1) but it is largely due to the lower stability (by 29.1 kJ mol−1) of 3-phenylpropyne compared with the more highly conjugated 1-phenylpropyne. This result emphasizes that RSEs should not be taken as an absolute measure of radical stability. They reflect the stability of a radical relative to that of its molecular precursor. Indeed, in the case of 1PPR vs. 3PPR, the difference in RSEs is dominated by differences in the stabilities of the molecular precursors. The deviation from additivity of the RSE (DARSE) for 1PPR is −11.4 kJ mol−1. The negative sign indicates an antagonistic interaction between the phenyl and vinyl substituents. This partly reflects a saturation effect and has been found generally in disubstituted methyl radicals with substituents of this type.8| Radical | Label | Structure | ΔEr | Precursor | ΔEm | BDE | RSE | DARSE |
|---|---|---|---|---|---|---|---|---|
| 1-Phenylpropargyl | 1PPR |
|
0 |
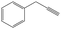
|
29.1 | 328.3 | 100.8 | −11.4 |
| 3-Phenylpropargyl | 3PPR |
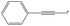
|
9.5 |
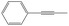
|
0 | 367.0 | 62.1 | — |
| trans-1-Vinylpropargyl | t-1VPR |
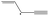
|
0 |

|
29.0 | 322.8 | 106.3 | −17.7 |

|

|
7.9 | 343.9 | 85.2 | — | |||
| 3-Vinylpropargyl | 3VPR |

|
10.0 |

|
0 | 361.8 | 67.2 | — |
| cis-1-Vinylpropargyl | c-1VPR |

|
1.6 |
|
26.8 | 326.6 | 102.5 | −21.5 |
|

|
6.5 | 346.8 | 82.2 | — | |||
| trans-1-Phenylallyl | t-1PAR |
|
0 |
|
18.1 | 323.4 | 105.6 | −24.7 |
|
|
0 | 341.5 | 87.6 | — | |||
| 2-Phenylallyl | 2PAR |
|
34.1 |
|
0.9 | 374.7 | 54.4 | — |
The vinylpropargyl radicals tell a similar story. The largest RSE (106.3 kJ mol−1) is found for the trans-1-vinylpropargyl radical (1VPR) and arises partly because 1VPR is the most stable radical and partly because it has the least stable precursor, 3-vinylpropyne (or pent-4-enyne). The DARSE for 1VPR is again negative (−17.7kJ mol−1). The energy difference between the cis and trans isomers, c-1VPR and t-1VPR, is just 1.6 kJ mol−1.
The trans-1-phenylallyl radical (t-1PAR) lies lower in energy than the 2-phenylallyl radical by 34.1 kJ mol−1. If 1-phenylpropene (trans-β-methylstyrene) is taken as the parent, then the existing conjugation between the olefinic and aromatic groups tends to stabilize the parent, which leads to an only moderate-sized RSE (87.6 kJ mol−1). On the other hand, if 3-phenylpropene is taken as the parent, then the RSE for t-1PAR is much larger at 105.6 kJ mol−1. The DARSE for t-1PAR (−24.7 kJ mol−1) is the largest of all the systems examined (Table 5).
The corollary of a large RSE is a low BDE. The BDEs of the molecular precursors of the radicals so far identified in our laboratory are in the 320 kJ mol−1 range, just three quarters of the BDE of methane. Spectroscopically, this equates to photon energies of 27000 cm−1, just outside the visible region, which suggests that predissociation may occur for the lowest electronic transitions of these species, which are in the UV. Furthermore, the lability of the hydrogens that dissociate to give rise to RSRs should engender deuteration enrichment at the radical site in natural samples of carbonaceous matter. This has been observed by ESR spectroscopy of RSRs extant in the Orgueille and Murchison meteorites, known to be rich in presolar carbonaceous material, the provenance of which is the aromatic carbonaceous material that pervades the interstellar medium.32–34
The diffuse interstellar bands (DIBs) are a series of absorption features exhibited in astronomical spectra of stars reddened by the diffuse interstellar medium, and are widely suspected to be carried by gas-phase, carbonaceous molecules.35 However, despite the certain existence and importance of RSRs in astronomical carbonaceous material, and despite these RSRs absorbing in the visible range, no DIB can yet be explained by the spectroscopy of one of these resonance-stabilized radicals.
Conclusions
We have identified two resonance-stabilized radicals in a molecular beam by synergistic application of several spectroscopic and computational techniques. The trans-1-phenylallyl (cinnamyl) radical was found to absorb with a strong origin transition at 19208 cm−1, and exhibited weak fluorescence with a lifetime of 63 ns. The inden-2-ylmethyl radical was found to absorb strongly at 19365 cm−1 and fluoresced more strongly than 1-phenylallyl radical with a 120 ns lifetime. This enabled the assignment of all ground-state frequencies excluding C–H stretches, and some out-of-plane modes. Ground-state frequencies were well reproduced by B3-LYP/6-311++G(d,p) scaled by 0.97. Vertical excitation energies were calculated to exceed the observed origin-band positions by ∼1000 cm−1.Vertical ionization energies calculated using B3-LYP/6-311++G(3df,3pd) and G3X(MP2)-RAD were found generally to underestimate and overestimate, respectively, the experimental values for a series of resonance-stabilized radicals. For the current systems, this generally leads to bracketing of the experimental IEs by the theoretical values, which was of great utility in confirming the identity of the radicals.
High-level G3X(MP2)-RAD calculations for the heterodisubstituted radicals 1-phenylpropargyl, 1-vinylpropargyl and 1-phenylallyl show that these radicals all have radical stabilization energies (RSEs) greater than 100 kJ mol−1. It was found that the larger positive RSEs compared with isomeric radicals arise partly because of relative stabilization of the radicals but largely because of relative destabilization of the closed-shell precursors. Deviations from additivity of the RSEs (DARSEs) are negative for all the disubstituted radicals examined here, indicating an antagonistic interaction between the substituents with respect to RSEs, and are greatest for the vinyl-substituted radicals.
Finally, the calculated BDEs for the disubstituted molecular precursors are low (approximately 320 kJ mol−1), and this has implications regarding the possible predissociation of these species.
Acknowledgements
This research was supported under the Australian Research Council's Discovery funding scheme (Project Numbers DP0985767, DP1095821 and CE0561607). T. P. T. acknowledges the University of Sydney for a University Postgraduate Award. N. C. acknowledges the Endeavor International Postgraduate Research Scholarship and the University of Sydney International Scholarship. A. S. M. thanks the University of Sydney for a Henry Bertie Florence Mabel Gritton Scholarship and thanks Professor Thomas Bally for hosting her visit to the University of Fribourg. K. N. thanks the Australian Research Council for the award of an Australian Research Fellowship. B. F. acknowledges the award of a Feodor Lynen Fellowship from the Alexander von Humboldt Foundation. L. R. thanks the ARC for an Australian Professorial Fellowship, and acknowledges funding from the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology, and generous allocations of computer time from the National Computational Infrastructure (NCI) National Facility and Intersect Australia Ltd.References
- C. S. McEnally, L. D. Pfefferle, B. Atakan and K. Kohse-Höinghaus, Prog. Energy Combust. Sci., 2006, 32, 247–294 CrossRef CAS
.
- H. B. Niemann, S. K. Atreya, J. E. Demick, D. Gautier, J. A. Haberman, D. N. Harpold, W. T. Kasprzak, J. I. Lunine, T. C. Owen and F. Raulin, J. Geophys. Res., 2010, 115, E12006 CrossRef
- T. P. Snow and V. M. Bierbaum, Annu. Rev. Anal. Chem., 2008, 1, 229–259 CrossRef CAS
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2001, 105, 7254–7266 CrossRef CAS
- L. B. Harding, S. J. Klippenstein and Y. Georgievskii, J. Phys. Chem. A, 2007, 111, 3789–3801 CrossRef CAS
- D. J. Henry, C. J. Parkinson, P. M. Mayer and L. Radom, J. Phys. Chem. A, 2001, 105, 6750–6756 CrossRef CAS
- M. L. Coote, C. Y. Lin, A. L. Beckwith and A. A. Zavitsas, Phys. Chem. Chem. Phys., 2010, 12, 9597–9610 RSC
- A. S. Menon, D. J. Henry, T. Bally and L. Radom, Org. Biomol. Chem., 2011, 9, 3636–3657 CAS
-
Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed
- A. S. Menon, G. P. F. Wood, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 13638–13644 CrossRef CAS
- N. J. Reilly, D. L. Kokkin, M. Nakajima, K. Nauta, S. H. Kable and T. W. Schmidt, J. Am. Chem. Soc., 2008, 130, 3137–3142 CrossRef CAS
- N. J. Reilly, M. Nakajima, B. A. Gibson, T. W. Schmidt and S. H. Kable, J. Chem. Phys., 2009, 130, 144313 CrossRef
- N. J. Reilly, M. Nakajima, T. P. Troy, N. Chalyavi, K. A. Duncan, K. Nauta, S. H. Kable and T. W. Schmidt, J. Am. Chem. Soc., 2009, 131, 13423–13429 CrossRef CAS
- Y. Ohshima and Y. Endo, J. Mol. Spectrosc., 1992, 153, 627 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C. 02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A. 1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed
-
H.-J. Werner, P. J. Knowles, F. R. Manby, M. Schtz, P. Celani, G. Knizia, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Kppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nickla, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO version 2008.1, University of Birmingham, Birmingham, UK, 2008 Search PubMed
- D. J. Henry, M. B. Sullivan and L. Radom, J. Chem. Phys., 2003, 118, 4849–4860 CrossRef CAS
- J. A. Sebree, V. V. Kislov, A. M. Mebel and T. S. Zwier, J. Phys. Chem. A, 2010, 114, 6255–6262 CrossRef CAS
- T. Izumida, K. Inoue, S. Noda and H. Yoshida, Bull. Chem. Soc. Jpn., 1981, 54, 2517–2518 CrossRef CAS
- M. Fukushima and K. Obi, J. Chem. Phys., 1992, 96, 4224–4232 CrossRef CAS
- P. Hemberger, M. Lang, B. Noller, I. Fischer, C. Alcaraz, B. K. C. de Miranda, G. A. Garcia and H. Soldi-Lose, J. Phys. Chem. A, 2011, 115, 2225–2230 CrossRef CAS
- X. Xing, B. Reed, K.-C. Lau, C. Y. Ng, X. Zhang and G. B. Ellison, J. Chem. Phys., 2007, 126, 171101 CrossRef
- B. Yang, Y. Li, L. Wei, C. Huang, J. Wang, Z. Tian, R. Yang, L. Sheng, Y. Zhang and F. Qi, Proc. Combust. Inst., 2007, 31, 555–563 CrossRef
- G. C. Eiden, F. Weinhold and J. C. Weisshaar, J. Chem. Phys., 1991, 95, 8665–8668 CrossRef CAS
- P. Hemberger, M. Steinbauer, M. Schneider, I. Fischer, M. Johnson, A. Bodi and T. Gerber, J. Phys. Chem. A, 2010, 114, 4698–4703 CrossRef CAS
- T. P. Troy, M. Nakajima, N. Chalyavi, R. G. C. R. Clady, K. Nauta, S. H. Kable and T. W. Schmidt, J. Phys. Chem. A, 2009, 113, 10279–10283 CrossRef CAS
- M. Tsuge, S. Hamatani, A. Kawai, K. Tsuji and K. Shibuya, Phys. Chem. Chem. Phys., 2006, 8, 256–263 RSC
- T. T. Nguyen and N. Sheppard, J. Catal., 1981, 67, 402–409 CrossRef CAS
- W. E. Sinclair, H. Yu, D. Phillips, R. D. Gordon, J. M. Hollas, S. Klee and G. Mellau, J. Phys. Chem., 1995, 99, 4386–4396 CrossRef CAS
- D. Gourier, F. Robert, O. Delpoux, L. Binet, H. Vezin, A. Moissette and S. Derenne, Geochim. Cosmochim. Acta, 2008, 72, 1914–1923 CrossRef CAS
- L. Binet, D. Gourier, S. Derenne, F. Robert and I. Coifini, Geochim. Cosmochim. Acta, 2004, 68, 881–891 CrossRef CAS
- O. Delpoux, D. Gourier, H. Vezin, L. Binet, S. Derenne and F. Robert, Geochim. Cosmochim. Acta, 2011, 75, 326–336 CrossRef CAS
- P. J. Sarre, J. Mol. Spectrosc., 2006, 238, 1–10 CrossRef CAS
- J. A. Sebree, N. M. Kidwell, E. G. Buchanan, M. Z. Zgierski and T. S. Zwier, Chem. Sci., 2011, 2 10.1039/c1sc00246e
Footnote |
| † Electronic Supplementary Information (ESI) available: Optimized geometries and calculated frequency tables for 2MIR, I2MR, t-1PAR and c-1PAR. See DOI: 10.1039/c1sc00247c/ |
| This journal is © The Royal Society of Chemistry 2011 |