Nickel-catalyzed amination of aryl carbamates and sequential site-selective cross-couplings†
Tehetena
Mesganaw
,
Amanda L.
Silberstein
,
Stephen D.
Ramgren
,
Noah F. Fine
Nathel
,
Xin
Hong
,
Peng
Liu
and
Neil K.
Garg
*
Department of Chemistry and Biochemistry, University of California, 607 Charles Young Drive East, Box 951569, Los Angeles, CA 90095, USA. E-mail: neilgarg@chem.ucla.edu
First published on 9th June 2011
Abstract
We report the amination of aryl carbamates using nickel-catalysis. The methodology is broad in scope with respect to both coupling partners and delivers aminated products in synthetically useful yields. Computational studies provide the full catalytic cycle of this transformation, and suggest that reductive elimination is the rate-determining step. Given that carbamates are easy to prepare, robust, inert to Pd-catalysis, and useful for arene functionalization, these substrates are particularly attractive partners for use in synthesis. The sequential use of carbamate functionalization/site-selective cross-coupling processes highlights the utility of this methodology.
Introduction
The discovery of methods for the assembly of carbon–nitrogen bonds continues to be an active area of research. Among the numerous tactics available for C–N bond formation, transition metal-catalyzed processes, led by Buchwald and Hartwig, have become some of the most widely used methods in chemical synthesis.1 Recent efforts have focused on the catalytic amination of phenol derivatives,2–5 as phenols are readily available, with certain analogs being ideally poised for the synthesis of polysubstituted arenes.6One particularly attractive class of electrophilies is the N,N-dialkyl aryl O-carbamate (1, Fig. 1). Features of these substrates include their ease of preparation,7 pronounced stability, and low reactivity toward Pd(0). Furthermore, aryl carbamates can be used for arene functionalization8 prior to a cross-coupling event, using either electrophilic aromatic substitution,9 directed o-metallation,10 or recently described Pd- or Ir-catalyzed methods.11
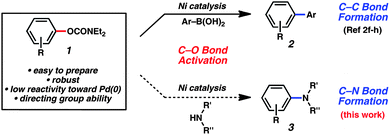 | ||
| Fig. 1 Known C–C and proposed C–N bond formation reactions using aryl carbamates as substrates. | ||
Although aryl carbamates have been employed in C–C bond forming processes (Fig. 1, 1 → 2),12,13 their use in amination reactions (1 → 3), has been less explored. Specifically, during the course of our own studies, only a single example of carbamate amination was reported using the N,N-diethylcarbamate derivative of phenol.4 Considering the importance of transition metal-catalyzed amination reactions in modern synthetic chemistry,1 coupled with the salient features of carbamate electrophiles, we sought to develop a general method for carbamate amination. In this manuscript, we report the broad scope of carbamate amination methodology, as well as a computational study of the full catalytic cycle. In addition, we demonstrate the value of these reaction partners for the synthesis of polysubstituted aryl amines using sequential carbamate functionalization/site-selective cross-coupling methodologies.
Results and discussion
Optimization and substrate scope
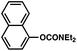
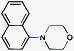
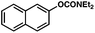
To initiate studies, we attempted the amination of diethylnaphthylcarbamates with morpholine under a variety of reaction conditions. Although Ni/PCy3-based conditions have been useful for achieving C–C bond formation, it was not possible to achieve amination using related procedures.14 After conducting an extensive survey of reaction parameters (e.g., nickel catalysts, ligands, solvents, bases, temperature, etc.) it was observed that combinations of Ni catalysts and N-heterocyclic carbene ligands promoted the desired amination. Our laboratory and Chatani's have previously noted analogous findings in couplings of sulfamates and pivalates, respectively.4,5We identified the use of catalytic Ni(cod)2, SIPr·HCl (4),15 and NaOtBu, in dioxane at 80 °C as optimal reaction conditions for amination and investigated the carbamate substrate scope (Table 1).16,17Naphthyl carbamates, which typically function well in the Suzuki–Miyaura coupling, were excellent substrates for the amination (entries 1 and 2). Non-fused aromatics were also tolerated by the methodology (entries 3–7). The electron-donating methoxy group (entry 4) and the electron-withdrawing trifluoromethyl group (entry 5) were suitable substrates. Methyl substituents at the para and meta positions were tolerated as well (entries 6 and 7).

| Entry | Ar-OCONEt2 | Product | Yieldb |
|---|---|---|---|
| a Conditions unless otherwise stated: Ni(cod)2 (5 mol%), 4 (10 mol%), carbamate substrate (1 equiv), morpholine (1.2 equiv), NaOtBu (1.4 equiv), 3 h. b Isolated yields. c Ni(cod)2 (15 mol%), 4 (30 mol%), morpholine (1.8 equiv), NaOtBu (2.2 equiv). d Ni(cod)2 (10 mol%), 4 (20 mol%). | |||
| 1 |
|
|
91% |
| 2 |
|
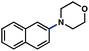
|
92% |
| 3 |
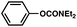
|

|
90% |
| 4c |
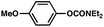
|

|
74% |
| 5 |
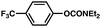
|

|
90% |
| 6d |
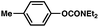
|

|
77% |
| 7 |

|
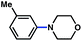
|
80% |
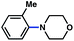
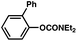
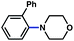
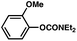
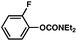
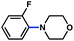
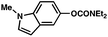
The feasibility of coupling o-substituted carbamates, in addition to heterocyclic substrates, was examined (Table 2).17 Of note, o-substituted aryl carbamates are readily accessible by functionalization of the parent carbamate (using directed metallation10 or transition metal-catalyzed processes11), but have proven to be exceptionally challenging substrates in the recently discovered nickel-catalyzed Suzuki–Miyaura coupling.12 We were delighted to find that a range of o-substituted phenylcarbamates could be employed in our amination methodology.18Carbon substituents were well-tolerated (entries 1 and 2), as were heteroatoms (entries 3–5). Furthermore, heterocyclic substrates containing indole or pyridine underwent coupling with morpholine under nickel catalysis (entries 6 and 7).
| Entry | Ar-OCONEt2 | Product | Yieldb |
|---|---|---|---|
| a Conditions unless otherwise stated: Ni(cod)2 (5 mol%), 4 (10 mol%), carbamate substrate (1 equiv), morpholine (1.2 equiv), NaOtBu (1.4 equiv), 3 h. b Isolated yields. c Ni(cod)2 (15 mol%), 4 (30 mol%), morpholine (1.8 equiv), NaOtBu (2.2 equiv). d Ni(cod)2 (15 mol%), 4 (30 mol%), morpholine (2.4 equiv), NaOtBu (2.2 equiv). e Ni(cod)2 (20 mol%), 4 (40 mol%), morpholine (1.2 equiv), NaOtBu (1.7 equiv), 120 °C. f Ni(cod)2 (10 mol%), 4 (20 mol%). g Ni(cod)2 (20 mol%), 4 (40 mol%), morpholine (1.8 equiv), NaOtBu (2.2 equiv). | |||
| 1c |
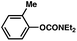
|
|
65% |
| 2d |
|
|
53% |
| 3e |
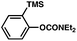
|

|
61% |
| 4d |
|

|
55% |
| 5f |
|
|
84% |
| 6g |
|
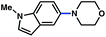
|
55% |
| 7 |
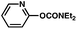
|
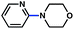
|
78% |
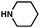
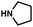
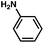
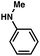
As shown in Table 3, a variety of amines can be employed in the carbamate amination.17,19 Both cyclic and acyclic secondary amines were tolerated (entries 1–3), in addition to anilines (entries 4–6). Of note, use of the sterically congested 2,6-dimethylaniline delivered the corresponding aminated product in 92% yield (entry 6). The methodology also allows for the coupling of amines with appended heterocycles (entries 7 and 8).
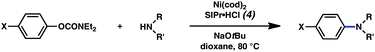
| Entry | Amine | Product | Yieldb |
|---|---|---|---|
| a Conditions unless otherwise stated: Ni(cod)2 (5 mol%), 4 (10 mol%), carbamate substrate (1 equiv), amine (1.2 equiv), NaOtBu (1.4 equiv), 3 h. b Isolated yields. c Ni(cod)2 (10 mol%), 4 (20 mol%). d Ni(cod)2 (15 mol%), 4 (30 mol%), amine (1.8 equiv), NaOtBu (2.2 equiv). e Ni(cod)2 (15 mol%), 4 (30 mol%), amine (2.4 equiv), NaOtBu (2.2 equiv). | |||
| 1c |
|

|
96% |
| 2 |
|

|
91% |
| 3 |
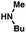
|
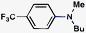
|
86% |
| 4d |
|
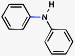
|
84% |
| 5d |
|
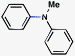
|
70% |
| 6e |
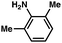
|
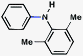
|
92% |
| 7c |
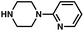
|

|
94% |
| 8c |
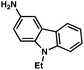
|
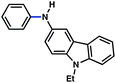
|
93% |
Computational studies
Although the mechanism of palladium-based aminations has been studied computationally,20–22 no theoretical studies of nickel-catalyzed aminations have been reported. Furthermore, computational studies involving unconventional phenol-based electrophiles (e.g., esters, carbamates, sulfamates) are rare and have only been examined in the context of C–C bond formation.23Accordingly, we conducted a DFT study of the nickel-catalyzed carbamate amination, using N,N-dimethylphenylcarbamate and dimethylamine as substrates.24The results of this computational study are shown in Fig. 2 in the form of a Gibbs free energy diagram, which in turn, provides the full catalytic cycle for carbamate amination. Analogous to Pd-catalyzed amination, three fundamental steps occur: oxidative addition, deprotonation, and reductive elimination.25 Previous mechanistic and theoretical studies on similar Pd- and Ni-catalyzed reactions suggested that the oxidative addition initiates via monoligated η2 complex 5.21,26,27 The oxidative addition occurs through five-centered transition state TS6, in which the carbonyl oxygen in the carbamate is coordinated with Ni.28 The electron-rich NHC ligand facilitates the oxidative addition, which requires only 5.7 kcal mol−1 with respect to the η2 complex 5.29 Similar oxidative additions with phosphine ligands require much higher activation energies (ΔG‡ = 13.5 kcal mol−1 when PCy3 ligand is used).23b
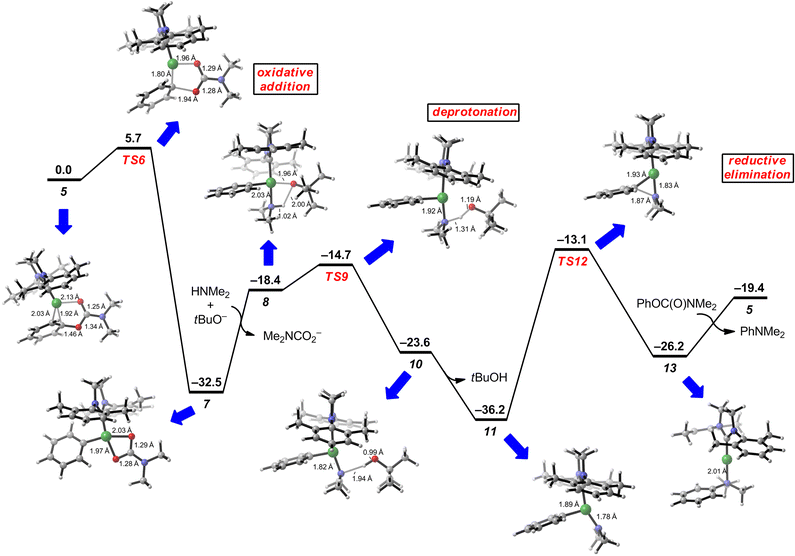 | ||
| Fig. 2 Gibbs free energy diagram of Ni-catalyzed amination of N,N-dimethylphenylcarbamate and dimethylamine. Energies are given in kcal mol−1. | ||
The oxidative addition leads to a stable intermediate (phenyl)nickel(II) carbamate intermediate 7 (–32.5 kcal mol−1). Complex 7 undergoes ligand exchange with dimethylamine and tert-butoxide to liberate carbamate anion and form intermediate 8 (–18.4 kcal mol−1). This ligand exchange process is endergonic, mainly due to entropic effects. The proton transfer from the coordinated amine to tert-butoxide (TS9) requires only 3.7 kcal mol−1activation energy from complex 8.30 Subsequent dissociation of tert-butanol gives the (phenyl)(amino)nickel(II) complex 11 (–36.2 kcal mol−1). Reductive elimination then occurs through TS12 (–13.1 kcal mol−1), which affords the product complex 13 (–26.2 kcal mol−1). The reductive elimination from 11 to TS12 requires 23.1 kcal mol−1 and is the rate-limiting step in the catalytic cycle. Thus, the overall energy span31 of the catalytic cycle is 23.1 kcal mol−1, in agreement with the experimental observations that the amination reaction readily occurs under slightly elevated temperatures. The barrier for reductive elimination with the Ni(NHC) catalyst is much higher compared to that of Pd-phosphine catalysts.20b,22e Following the reductive elimination, the reactant complex 5 can be regenerated by ligand exchange from the product complex 13 to initiate another catalytic cycle. The whole catalytic cycle is exergonic by 19.4 kcal mol−1.32
Site-selective cross-couplings and synthetic applications
Fig. 3 highlights a series of experiments that were undertaken to explore carbamate directing group ability and the low reactivity of these substrates to conventional catalytic transformations. The key substrate for our studies, dihydroquinone derivative 14, was selected with the aim of simultaneously probing the reactivity of aryl sulfamates, which have also proven to be extremely useful electrophiles in nickel-catalyzed couplings. Lithiation/bromination of substrate 14 provided trisubstituted arene 15. In accord with literature precedent by Snieckus,13f the lithiation proceeded selectively adjacent to the carbamate. Bromoarene 15 was subsequently employed in a series of C–C and C–heteroatom bond constructions. Pd-catalyzed arylation (15 → 17), alkylation (15 → 18), and amination (15 → 19) proceeded smoothly, as did Cu-catalyzed C–N bond formation (15 → 16). In all cases, the sulfamate and carbamate were not disturbed.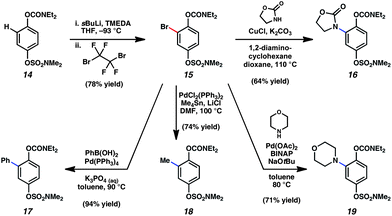 | ||
| Fig. 3 Carbamate functionalization and low reactivity of carbamates and sulfamates toward conventional Pd- and Cu-catalyzed couplings. | ||
Having demonstrated the robust nature of carbamates and sulfamates to a variety of conditions, we examined the subsequent cross-couplings of these functional groups (Fig. 4). o-Methylated derivative 18, prepared by either o-bromination/Stille coupling (see Fig. 3) or direct o-methylation of 14, was used in this study. We have found that the sulfamate of 18 is more reactive compared to the carbamate, and that high degrees of selectivity can be obtained in arylation.33 Suzuki–Miyaura coupling of 18 furnished carbamate 20 in 52% yield. Subsequently, carbamate 20 was employed in our nickel-catalyzed amination to furnish polysubstituted aryl amine 21. We expect that the ability to consecutively cross-couple bromides, sulfamates, and carbamates will be useful in the synthesis of complex molecules.
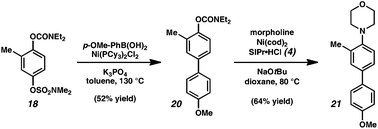 | ||
| Fig. 4 Synthesis of polysubstituted arenes using sequential sulfamate/carbamate couplings. | ||
Conclusions
In summary, we have found that aryl carbamates are excellent substrates for the nickel-catalyzed amination reaction. The scope of the methodology is broad with respect to both coupling partners, and includes the coupling of electron-rich, heterocyclic, and sterically congested carbamates. DFT calculations reveal the full catalytic cycle of the nickel-catalyzed carbamate amination and suggest that reductive elimination (23.1 kcal mol−1 barrier) is the rate-determining step. Moreover, we have demonstrated that aryl carbamates are outstanding precursors for the synthesis of polysubstituted aryl amines using sequential carbamate functionalization/site-selective coupling processes. The use of this methodology in natural product synthesis will be reported in due course.Acknowledgements
The authors are grateful to Boehringer Ingelheim, Bristol-Myers Squibb (T.M.), DuPont, Eli Lilly, Amgen, NOBCChE/GlaxoSmithKline (T.M.), the University of California, Los Angeles, and the Chemistry-Biology Interface training program (A.L.S., USPHS National Research Service Award GM08496) for financial support. We thank the Garcia-Garibay laboratory (UCLA) for access to instrumentation, Dr. John Greaves (UC Irvine) for mass spectra, K. N. Houk for helpful discussions, and Materia Inc. for chemicals.Notes and references
- (a) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS; (b) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS; (c) A. R. Muci and S. L. Buchwald, Practical Palladium Catalysts for C–N and C–O Bond Formation, in Topics in Current Chemistry, Vol. 219 (Ed: N. Miyaura), Springer–Verlag, Berlin, 2001, p 131 Search PubMed; (d) M. Kienle, S. R. Dubbaka, K. Brade and P. Knochel, Eur. J. Org. Chem., 2007, 4166–4176 CrossRef CAS; (e) J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS; (f) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- For examples of tosylate and mesylate amination, see: (a) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 7369–7370 CrossRef CAS; (b) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 8704–8705 CrossRef CAS; (c) T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 13848–13849 CrossRef CAS; (d) C.-Y. Gao and L.-M. Yang, J. Org. Chem., 2008, 73, 1624–1627 CrossRef CAS; (e) B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS; (f) C. M. So, Z. Zhou, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 6402–6406 CrossRef CAS.
- The amination of aryl methyl ethers is largely limited to the coupling of 2-methoxynaphthalene with cyclic secondary amines. Deviation from these substrates leads to significantly lower yields (ca. 30–50%). M. Tobisu, T. Shimasaki and N. Chatani, Chem. Lett., 2009, 38, 710–711 Search PubMed.
- For the amination of aryl pivalates, see: T. Shimasaki, M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2010, 49, 2929–2932 Search PubMed.
- For sulfamate amination, see: (a) S. D. Ramgren, A. L. Silberstein, Y. Yang and N. K. Garg, Angew. Chem., Int. Ed., 2011, 50, 2171–2173 Search PubMed; (b) L. Ackermann, R. Sandmann and W. Song, Org. Lett., 2011, 13, 1784–1786 Search PubMed.
- For recent reviews and highlights regarding the cross-coupling of phenolic derivatives, see: (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS; (b) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 1728–1759 CrossRef CAS; (c) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486–1495 CrossRef CAS; (d) C. E. I. Knappke and A. Jacobi von Wangelin, Angew. Chem. Int. Ed., 2010, 49, 3568–3570 CAS; (e) L. J. Goossen, K. Goossen and C. Stanciu, Angew. Chem., Int. Ed., 2009, 48, 3569–3571 Search PubMed.
- Aryl N,N-diethylcarbamates are readily prepared from aryl alcohols and commercially available N,N-diethylcarbamoyl chloride. The latter reagent costs approximately $20–25 per mol ($0.15–$0.20 per gram) from common chemical suppliers, such as Alfa Aesar and Aldrich Chemical Co., Inc..
- Other phenol-based amination partners cannot be used effectively for directed metallation/functionalization. Specifically, aryl sulfonates and pivalates are not suitable substrates, whereas aryl methyl ethers are poor metalation substrates. Aryl sulfamates can be used in ortho-lithiation, but the sulfamate directing group ability is approximately 20x less compared to carbamates; see ref. 13f.
- M. B. Smith and J. March, in March's Advanced Organic Chemistry, 6th ed., John Wiley & Sons, Inc., New Jersey, 2007, p 670 Search PubMed.
- (a) V. Snieckus, Chem. Rev., 1990, 90, 879–933 CrossRef CAS; (b) C. G. Hartung and V. Snieckus, in Modern Arene Chemistry (Ed.: D. Astruc), Wiley-VCH, New York, 2002, pp 330–367 Search PubMed; (c) T. Macklin and V. Snieckus, in Handbook of C–H Transformations (Ed.: G. Dyker), Wiley-VCH, New York, 2005, pp 106–119 Search PubMed.
- (a) R. B. Bedford, R. L. Webster and C. J. Mitchell, Org. Biomol. Chem., 2009, 7, 4853–4857 RSC; (b) X. Zhao, C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 5837–5844 CrossRef CAS; (c) T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 4978–4979 CrossRef CAS; (d) K. Yamazaki, S. Kawamorita, H. Ohmiya and M. Sawamura, Org. Lett., 2010, 12, 3978–3981 CrossRef CAS.
- For the use of aryl carbamates in C–C bond forming processes, see: (a) K. W. Quasdorf, M. Riener, K. V. Petrova and N. K. Garg, J. Am. Chem. Soc., 2009, 131, 1774–17749 Search PubMed; (b) A. Antoft-Finch, T. Blackburn and V. Snieckus, J. Am. Chem. Soc., 2009, 131, 17750–17752 CrossRef CAS; (c) L. Xi, B.-J. Li, Z.-H. Wu, X.-Y. Lu, B.-T. Guan, B.-Q. Wang, K.-Q. Zhao and Z.-J. Shi, Org. Lett., 2010, 12, 884–887 CrossRef CAS.
- For C–C bond forming processes involving aryl pivalates, methyl ethers, or sulfamates, see: (a) K. W. Quasdorf, X. Tian and N. K. Garg, J. Am. Chem. Soc., 2008, 130, 14422–14423 CrossRef CAS; (b) B.-T. Guan, Y. Wang, B.-J. Li, D.-G. Yu and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 14468–14470 CrossRef CAS; (c) B.-J. Li, Y.-Z. Li, X.-Y. Lu, J. Liu, B.-T. Guan and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 10124–10127 CrossRef CAS; (d) M. Tobisu, T. Shimasaki and N. Chatani, Angew. Chem., Int. Ed., 2008, 47, 4866–4869 CrossRef CAS; (e) J. W. Dankwardt, Angew. Chem., Int. Ed., 2004, 43, 2428–2432 CrossRef CAS; (f) T. K. Macklin and V. Snieckus, Org. Lett., 2005, 7, 2519–2522 Search PubMed; (g) P. M. Wehn and J. Du Bois, Org. Lett., 2005, 7, 4685–4688 CrossRef CAS.
- This finding is likely not surprising since C–N and C–C bond forming processes would proceed through fairly different mechanistic pathways (although the initial oxidative addition step would be common to both processes).
- SIPr·HCl (4), is commercially available (CAS# 258278-25-0).
- Although these conditions are broadly useful, aminations of certain substrates proceed even at ambient temperature. For example, N,N-diethylphenylcarbamate undergoes coupling with morpholine in 24 h at 23 °C using 5 mol% catalyst (∼90% conversion based on 1H NMR analysis with internal standard).
- For certain substrates, the use of standard reaction conditions led to slow conversion to aminated product. In these cases, higher catalyst, ligand, and/or amine loadings could be used to expedite reaction progress as indicated in Tables 1–3. The formation of undesired byproducts is not typically observed.
- The carbamate derived from 2,6-dimethylphenol fails to undergo amination under our reaction conditions.
- Attempts to effect the amination of primary aliphatic amines, such as benzyl amine and n-butylamine, led to the recovery of starting material, albeit with some non-specific decomposition.
- (a) For a pertinent review, see: L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692–1705 Search PubMed; (b) For a theoretical study on the Pd-catalyzed amination of bromobenzenes, see: J. N. Harvey, N. Fey and C. L. McMullin, J. Mol. Catal. A: Chem., 2010, 324, 48–55 Search PubMed.
- For theoretical studies on Pd-mediated oxidative addition, see: (a) M. Ahlquist, P. Fristrup, D. Tanner and P. O. Norrby, Organometallics, 2006, 25, 2066–2073 CrossRef CAS; (b) M. Ahlquist and P. O. Norrby, Organometallics, 2007, 26, 550–553 CrossRef CAS; (c) Z. Li, Y. Fu, Q. X. Guo and L. Liu, Organometallics, 2008, 27, 4043–4049 CrossRef CAS; (d) J. N. Harvey, N. Fey and J. Jover, J. Mol. Catal. A: Chem., 2010, 324, 39–47 Search PubMed; (e) C. L. McMullin, J. Jover, J. N. Harvey and N. Fey, Dalton Trans., 2010, 39, 10833–10836 RSC.
- For theoretical studies on Pd-mediated reductive elimination, see: (a) V. P. Ananikov, D. G. Musaev and K. Morokuma, J. Am. Chem. Soc., 2002, 124, 2839–2852 CrossRef CAS; (b) V. P. Ananikov, D. G. Musaev and K. Morokuma, Organometallics, 2005, 24, 715–723 CrossRef CAS; (c) E. Zuidema, P. W. N. M. van Leeuwen and C. Bo, Organometallics, 2005, 24, 3703–3710 CrossRef CAS; (d) V. P. Ananikov, D. G. Musaev and K. Morokuma, Eur. J. Inorg. Chem., 2007, 5390–5399 CrossRef CAS; (e) T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 12003–12010 CrossRef CAS; (f) T. Koizumi, A. Yamazaki and T. Yamamoto, Dalton Trans., 2008, 3949–3952 RSC; (g) A. Ariafard and B. F. Yates, J. Organomet. Chem., 2009, 694, 2075–2084 CrossRef CAS.
- (a) Z. Li, S.-L. Zhang, Y. Fu, Q.-X. Guo and L. Liu, J. Am. Chem. Soc., 2009, 131, 8815–8823 CrossRef CAS; (b) K. W. Quasdorf, A. Antoft-Finch, P. Liu, A. L. Silberstein, A. Komaromi, T. Blackburn, S. D. Ramgren, K. N. Houk, V. Snieckus and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 6352–6363 Search PubMed.
- All geometries were optimized using the B3LYP functional and a mixed basis set of SDD for Ni and 631G(d) for other atoms. A larger base set, SDD for Ni and 6-311 + G(2d, p) for other atoms was used for single point energy calculations and solvation energy corrections using the CPCM model. Single point calculations using the B3P86 and B3PW91 functionals were also performed and give comparable results to the B3LYP calculations (see ESI† for details) The isopropyl groups on the SIPr ligand were replaced with methyls in the calculations to reduce computational cost. All calculations were performed using Gaussian 09. Gaussian 09, revision B.01,M. J. Frischet al.Gaussian, Inc., Wallingford CT, 2010.
- The Ni-catalyzed amination of aryl carbamates is not inhibited in the presence of galvinoxyl radical or BHT; thus, we propose that a Ni(I)/Ni(III) catalytic cycle is likely not operative. For examples where additives such as galvinoxyl radical or BHT are used to probe the presence of Ni(I) intermediates, see: (a) T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS; (b) T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 7547–7560 CrossRef CAS; (c) J. Kochi, Pure Appl. Chem., 1980, 52, 571–605 CrossRef CAS; (d) B. H. Lipshutz, J. A. Sclafani and P. A. Blomgren, Tetrahedron, 2000, 56, 2139–2144 CrossRef CAS; (e) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS; (f) J. Zhou, Ph.D. Thesis, Massachusetts Institute of Technology, 2005; (g) V. B. Phapale, M. Guisan-Ceinos, E. Bunuel and D. J. Cardenas, Chem.–Eur. J., 2009, 15, 12681–12688 Search PubMed. For recent examples of catalytic reactions that proceed viaNi(I)/Ni(III) cycles, see: (h) K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546–2552 Search PubMed; (i) S. Miyazaki, Y. Koga, T. Matsumoto and K. Matsubara, Chem. Commun., 2010, 46, 1932–1934 RSC.
- The monoligated oxidative addition is suggested as a preferred pathway in most Pd- and Ni-catalyzed oxidative additions with phosphine and NHC ligands: (a) J. P. Stambuli, R. Kuwano and J. F. Hartwig, Angew. Chem., Int. Ed., 2002, 41, 4746–4748 CrossRef CAS; (b) J. P. Stambuli, M. Buehl and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 9346–9347 CrossRef CAS; (c) J. P. Stambuli, C. D. Incarvito, M. Buehl and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 1184–1194 CrossRef CAS; (d) M. Yamashita and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 5344–5345 CrossRef CAS; (e) J. C. Green and B. J. Herbert, J. Organomet. Chem., 2005, 690, 6054–6067 CrossRef CAS; (f) L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, Organometallics, 2006, 25, 54–67 CrossRef CAS; (g) A. G. Sergeev, A. Zapf, A. Spannenberg and M. Beller, Organometallics, 2008, 27, 297–300 CrossRef CAS; (h) F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8141–8154 CrossRef CAS; (i) V. P. W. Böhm, C. W. K. Gstöttmayr, T. Weskamp and W. A. Herrmann, Angew. Chem., Int. Ed., 2001, 40, 3387–3389 CrossRef CAS.
- We calculate that Ni(NHC)2 is 22.6 kcal mol−1 more stable in solution compared to 5. However, increasing the concentration of the NHC ligand does not suppress the amination reaction (see ESI†), suggesting that Ni(NHC)2 is not the catalyst resting state. Although not presently well-understood, it is likely that the dissociation of one NHC ligand from Ni(NHC)2 is facilitated by other species in the reaction mixture. For similar hypotheses involving NHC dissociation from Ni(NHC)2 complexes, see: (a) P. M. Zimmerman, A. Paul, Z. Zhang and C. B. Musgrave, Angew. Chem., Int. Ed., 2009, 48, 2201–2205 CrossRef CAS; (b) P. M. Zimmerman, A. Paul and C. B. Musgrave, Inorg. Chem., 2009, 48, 5418–5433 CrossRef CAS.
- An alternative pathway involving three-centered oxidative addition transition state requires much higher activation energy than the five-centered pathway viaTS6. Similar effects are observed in oxidative additions of carbamates and sulfamates with Ni(PR3); see ref. 23b.
- Similar effects of ligands have been reported previously: (a) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS; (b) M. Ahlquist and P. Norrby, Organometallics, 2007, 26, 550–553 CrossRef CAS; (c) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS; (d) Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef CAS.
- Since the mechanism involves charged species, the energetics of the deprotonation step is expected to be strongly affected by solvation effects. We have tested a number of DFT methods and basis sets with the CPCM solvation model. These results (provided in the ESI†) all indicate that deprotonation is not the rate-limiting step..
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS.
- The energy difference (−19.4 kcal mol−1) between complex 5 at the beginning and end of the plot represents the energy released in the catalytic cycle. ref. 31.
- Competition experiments between phenol-derived carbamates and sulfamates indicate that sulfamates are inherently more reactive than carbamates in both the nickel-catalyzed Suzuki–Miyaura coupling and amination. The preference for sulfamate coupling seen in the conversion of 18 → 20 is likely heightened because of the carbamate's ortho substituent.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00230a |
| This journal is © The Royal Society of Chemistry 2011 |