Salts dissolved in salts: ionic liquid mixtures†
Matthew Y.
Lui
,
Lorna
Crowhurst
,
Jason P.
Hallett
*,
Patricia A.
Hunt
,
Heiko
Niedermeyer
and
Tom
Welton
*
Department of Chemistry, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: j.hallett@imperial.ac.uk; Tel: +44 0207 594 3992t.welton@imperial.ac.uk; Tel: +44 0207 594 5763
First published on 2nd June 2011
Abstract
Solvents and solutions are ubiquitous in chemistry. For instance, in synthesis the solvent allows reagents to mix intimately so that reactions between these may occur. Consequently, understanding how solutes behave in solutions has been one of the major themes of chemistry throughout its history. Ionic liquids (liquid salts) are an exciting recent addition to the range of available solvents. Here we show that these solvents interact with dissolved salts to give solutions that are completely different from those of salts in either traditional organic solvents or water. Observations of these ideal salt solutions will require new models of solvation and polarity and have the potential to lead to new chemical processes.
Introduction
Modern theories of solvation have been formulated to understand observations of the behaviours of solutes in molecular solvents.1 For molecular solutes at high dilution we generally see polarity dependent solvation (molecule–molecule interactions) of the individual molecules. For dissolved salts a number of solute species ranging from fully dissociated solvated free ions (molecule–ion interactions) to contact ion pairs (molecule–ion–ion–molecule) are observed.2 The introduction of ionic liquids as useful solvents for the chemical industry3 is requiring that these observations and theories are expanded to encompass these new materials. To date, investigations of how ionic liquids interact with dissolved species have concentrated upon molecular solutes (molecule–ion interactions).4 Recently, our attention has been drawn to the study of dissolved salts in ionic liquids, which is showing some remarkable results arising from the exclusively ion–ion interactions that are unique to these molecule-free solutions.Ionic liquids are liquids composed entirely of ions when pure. Commonplace salts melt at high temperatures, such as NaCl at 801 °C.5 However, it is possible to prepare ionic liquids at room temperature by combining a large, asymmetric organic cation with a bulky, charge diffuse anion (see Fig. 1). These low-melting ionic liquids have been the subject of much interest in recent years.6
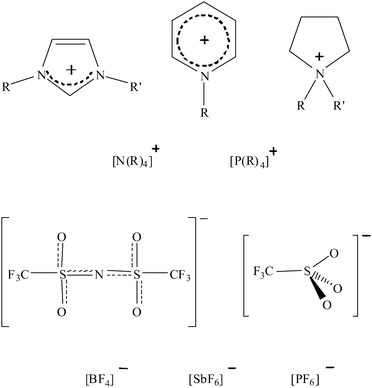 | ||
| Fig. 1 Common ions used in ionic liquids. | ||
Intuitively, one would expect a liquid composed entirely of ions to be highly polar. Indeed, studies of reaction rates7 and spectra of some solute species4,8 have indicated that ionic liquids behave similarly to polar molecular liquids. However, other spectroscopic results9,10 and the dielectric spectroscopy measurements by Weingärtner et al.11 have shown that some of the same ionic liquids have static dielectric constants as low as 10–15, similar to non-polar molecular liquids. The discrepancy between these low measurements of bulk solvent polarity and the typical effects of ionic liquids on reactions (generally indicating high polarity) has formed a barrier to the proper understanding of the interactions that dominate ionic liquid solutions.
Previously we have proposed that the reactivity resulting from mixing two different and reactive salts (charged electrophiles and nucleophiles) together is highly dependent on the type of solvent, with molecular and ionic liquids exhibiting fundamentally different reaction pathways. In ionic liquids, the salts behaved as discrete reactive species, whereas in molecular solvents neutral clusters of ions are formed as the reactive species.12 This was the first report of any reaction phenomenon unique to ionic liquids, and we proposed that this was due to the complete lack of ion-pairing of these solute salts in ionic liquids. This hypothesis has subsequently been supported by theoretical modelling,13 but we have been searching for further experimental evidence to support it and to also resolve the conflicting reports of ionic liquid polarity. This lack of ion-pairing indicates a very polar solvent in which the solvent ion–solute ion interactions are sufficient to break the strong attractive Coulombic interactions of any solute ion pairs.
Kosower's Z-scale was one of the first successful empirical polarity scales and is based upon the position of the absorption maximum of the longest wavelength charge-transfer band of 1-ethyl-4-(methoxycarbonyl)pyridinium iodide, [Py]I.14 In solution, this salt is only spectroscopically active when its ions are in direct contact, so allowing charge transfer to occur. Therefore, the intensity of this band is expected to be a good indicator of the number of [Py]I contact ion pairs in solution and can be used as a probe for the amount of ion-pairing in low and high polarity liquids. Bagchi,15 and Hemmes and co-workers16 both reported evidence for the presence of solvent-separated ion pairs (an ion pair with a single molecule of solvent between the ions, SSIP) in the molecular solutions of N-alkylpyridinium iodide. Binder et al. also showed the presence of solvent separated ion-pairs in solutions of 1-alkyl-(4-cyanopyridinium) iodide.17 Since a solvent separated ion pair cannot lead to charge transfer, both of the following equilibria must be considered for the closely related Kosower's salt, as in Scheme 1.
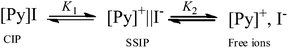 | ||
| Scheme 1 | ||
Since Kosower's salt in solution can exist as a contact ion pair, a solvent separated ion pair, or separate discrete solvated ions (Scheme 1), the concentration of spectroscopically active contact ion pairs (CIP) can be related to the total amount of Kosower's salt added (C0, see supplementary information for derivation):
| (1 + K1)CCIP + K0.51K0.52C0.5CIP − C0 = 0 | (1) |
Experimentally, the absorbance (A) of a spectroscopically active species is proportional to the concentration of that species in solution via the Beer–Lambert law (eqn (2)), where ε is the molar extinction coefficient, and l is the path length of the spectrophotometric cell. For most molecular solutes, ε is constant across a very wide concentration range, as the absorbance of an isolated molecule is not dependent upon its concentration.
| A = εCl | (2) |
In using Kosower's salt, only the contact ion pairs will give rise to absorbance and hence the total concentration of the salt added (C0) can differ from the concentration of the contact ion pairs CCIP, and it is only the concentration of contact ion pairs that is directly proportional to the absorbance of the charge transfer band. For an individual measurement, however, one can calculate an extinction coefficient, εapp, (not a molar absorptivity of CIP, because the concentration of these is unknown) from the ratio of the absorbance to the total concentration of salt. In low polarity molecular solvents, such as chloroform, the observed extinction coefficient εapp can reach values as high as 1400 L mol−1 cm−1. In high polarity solvents, such as methanol, this value drops to 30 L mol−1 cm−1.14 This indicates a higher concentration of contact ion pairs in the low polarity solvent and a lower concentration of ion pairs in the high polarity (or more dissociating) solvent, for any given amount of added Kosower's salt. This is usually well modelled using the liquid's dielectric constant.14 Here, we report the ion association/dissociation behaviour of 1-ethyl-4-(methoxycarbonyl)pyridinium iodide in a number of ionic and molecular liquids and with the aid of mathematical and computational modelling provide insight into the polarity of and extent of ion-pairing in ionic liquids.
Results
The first point of note is that we do observe a spectrum for Kosower's salt in all of the ionic liquids that we tested. This is in complete contradiction to our expectation from our previous kinetics results. The positions of the absorption maxima of these spectra yield the Z-values. In molecular liquids, the Z-values depend somewhat upon the salt concentration – the lower Z, the greater its sensitivity to concentration.14 For instance, a substantial change was observed in 1,2-dichloroethane in which the Z-values changed from 63.3 kcal mol−1 at 0.2 mM to 65.1 kcal mol−1 at 20 mM. No, or small, effects were found in more polar liquids (e.g. in 1-butanol and acetonitrile), but overall Z and thus polarity only increase with concentration in molecular solvents. In contrast, the Z-values of the ionic liquids show a small, but perceptible decrease when more Kosower's salt is added. Consequently, comparisons of Z-values for different solvents must be made at a fixed concentration.Our common ionic liquids have Z-values in the range of 72.7–76.5 (Table 1), which is generally lower than those of polar protic liquids (e.g. 1-butanol) and higher than polar non-hydrogen-bonding liquids (e.g. acetonitrile). A Regression analysis carried out on the Z data found to correlate well with Kamlet–Taft solvatochromic parameters α (hydrogen bond donation ability), β (hydrogen bond accepting ability) and π* (dipolarity/polarisability). Common ionic liquids have lower α than polar protic liquids but higher α than polar non-hydrogen-bonding liquids, hence their Z-values are in this range. Similar conclusions have been drawn from other studies of the polarities of ionic liquids, using molecular probes.20
| Liquid | Z | α | β | π* |
|---|---|---|---|---|
| a Ref. 18. b Ref. 19. | ||||
| Water | 94.6a | 1.16b | 0.50b | 1.13b |
| Methanol | 83.6a | 1.05b | 0.61b | 0.73b |
| Ethanol | 79.6a | 0.86a | 0.75a | 0.54a |
| Acetic acid | 79.2a | 1.12a | 0.45a | 0.64a |
| 1-butanol | 78.6 | 0.84 | 0.84 | 0.47 |
| [C4C1im][BF4] | 76.5 | 0.62 | 0.37 | 1.05 |
| [C4C1im][OTf] | 76.0 | 0.62 | 0.49 | 1.00 |
| [C4C1im][SbF6] | 75.8 | 0.62 | 0.15 | 1.04 |
| [C4C1im][NTf2] | 74.3 | 0.61 | 0.23 | 0.99 |
| [C4C1pyrr][NTf2] | 73.3 | 0.42 | 0.29 | 0.96 |
| [C4C1C1im][NTf2] | 72.7 | 0.38 | 0.26 | 1.02 |
| Propylene carbonate | 72.4 | 0.00 | 0.40 | 0.83 |
| Acetonitrile | 71.3a | 0.35b | 0.37b | 0.80b |
| DMSO | 70.2a | 0.00a | 0.76a | 1.00 |
| 1,2-dichloroethane | 65.2 | 0.00 | 0.10 | 0.81 |
Absorptivity
While the ionic liquids give unremarkable Z-values, the absorptivities of their solutions of Kosower's salt behave very differently to those of solutions of [Py]I in molecular solvents (see Fig. 2). For a sufficiently low polarity molecular liquid, a horizontal line with high εapp would be obtained if all of the ions were paired at all concentrations and, therefore, are spectroscopically active (C0 = CCIP). However, in both polar and non-polar molecular solvents, Kosower has shown that a curve is normally obtained.14 The straight lines obtained for this selection of ionic liquids are, therefore, highly unusual. The slopes (585, 682, 705, 759, 823, 849) increase in order of increasing ionic liquid molar volume, indicating the different ratios of solute to solvent ions in these solutions. According to Weingärtner,11 the dielectric constants of the ionic liquids used in this study lie in the range of 10–15, which is in between the values of 1,2-dichloroethane and 1-butanol. Our experimental results clearly indicate that the usual correlation found between the dielectric constants and degree of ion contact for molecular solvents2 does not hold for ionic liquids.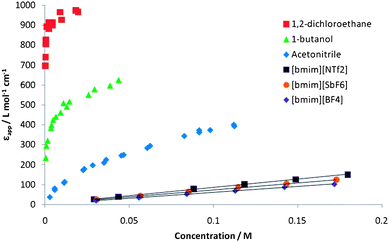 | ||
| Fig. 2 Apparent molar extinction coefficient, εapp, as a function of total concentration of 1-ethyl-4-(methoxycarbonyl)pyridinium iodide in various solvents. For clarity the results for only three of the ionic liquids are shown. For molecular solvents measurements were made up to the saturation concentration of Kosower's salt. | ||
The observation of a weak absorption band for solutions of Kosower's salt in ionic liquids clearly shows that some, but not all, of its pyridinium cations are in direct contact with iodide anions, but no model involving any equilibria between ion pairs and free ions can give rise to a linear dependence of εapp on total concentration. Consequently, some other model is required to explain these observations.
Liquid pseudo-lattice model
The experimentally observed linear relationship between εapp and total Kosower's salt concentration implies that the probability of a direct contact between ions of Kosower's salt is dependent only on the concentration of these ions. This behavior is indicative of statistically random ion contacts, rather than solvated ion pairs, i.e., there is no attractive nor repulsive interaction leading to solute ion pairing. This led us to utilize a random ionic structure based on a lattice site model with zero site exchange energy (Fig. 3). Using this model, at any concentration the statistical probability for the pyridinium and iodide ions to be located on adjacent lattice sites can be calculated. Given that r is the ratio of the total possible anion sites in the first solvation shell surrounding each pyridinium cation to the number of sites that give rise to an absorption, where mI = number of iodide ions in the mixture and mT = total number of anions in the mixture (calculated from the pure ionic liquid molar volume), then the probability of having at least one iodide anion in the solvent shell, P[Py]I, is (see supplementary information for derivation):| P[Py]I = 1 − [(mT − mI)/mT]r | (3) |
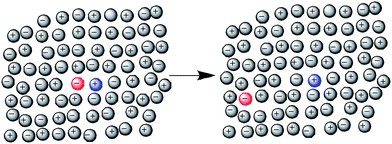 | ||
| Fig. 3 Schematic of Kosower's salt solvated as a contact ion pair (left) or as separate individual ions (right). | ||
The absorbance will now be proportional to P[Py]I in accordance with the Beer–Lambert law. Curves for the concentration-dependence of the molar absorptivity for various numbers, n, of ions in the cybotactic shell of the pyridinium ion have been calculated and are shown in Fig. 4 for the ionic liquid [C4C1im][NTf2]. For r greater than 1 this equation is a power law, however it is clear from the linear relation at the experimentally realistic concentrations shown in Fig. 4 we remain in the linear regime. Thus, this lattice site model is consistent with our experimental results.
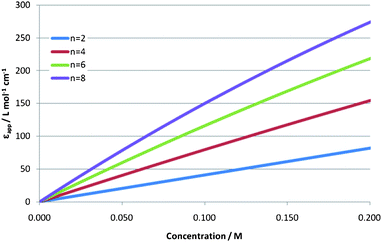 | ||
| Fig. 4 Theoretical apparent molar extinction coefficient, εapp, as a function of total concentration of charge-transfer salt, C0, based on eqn (1). n is the number of ions in the cybotactic shell of a solute. | ||
The slopes of these lines are expected to be proportional to the molar volume of the ionic liquid employed, with larger ionic liquids yielding larger slopes. This is also consistent with our experimental findings. The value of this slope can therefore be used to estimate the number of anions in the cybotactic shell of the pyridinium cation. After correcting for the molar volume of the ionic liquid employed, the slopes quoted above all collapse to a single line giving a value of r ≈ 2. Given that it has been shown that there are two sites around the pyridinium cation in which the iodide can give charge transfer (one above and one below the plane of the ring)14n ≈ 4. This value is lower than the value found by Hardacre using neutron diffraction data, for the 1,3-dimethylimidazolium cation in ionic liquid 1,3-dimethylimidazolium chloride, which indicated a first solvation shell of 6 anions.21 This probably arises because of the differences in the relative sizes of the cations and anions in the different systems, as Hardacre subsequently found that less rigid ordering exists when the larger [NTf2] anion replaced chloride.22
The fact that the charge-transfer behaviour in ionic liquids followed this pseudo-lattice model indicates that the interactions of the different ionic species (cations and anions deriving from either the ionic liquid or the Kosower's salt) with their immediate surroundings have very similar energies. Thus, there is a near-zero energy penalty to site exchange between ions and the solute–solute, solute–solvent and solvent–solvent interactions are indistinguishable. This is quite unlike the situations found in solutions of salts in molecular solvents, whether of high or low polarity, and is much more akin to the situation found in mixtures of similarly structured molecular liquids (e.g. the nearly ideal mixtures of toluene and benzene). In ionic liquid solvents, solute ions are neither held together in classical solvated contact ion pairs nor kept apart as solvated free ions. Thus, it appears to us that a unique solvation paradigm can exist for salts dissolved in an ionic solvent – the solute behaves as two distinct species, a cation and an anion, completely divorced from each other (highly screened) and capable of independently interacting with the solvent ions or other solute ions. Thus, these ionic liquids appear to form ideal mixtures of ions with the dissolved Kosower's salt.
These apparently ideal salt mixtures contrast with common observations that salts such as NaCl are insoluble in many ILs.6 In the case of NaCl, the very similar sizes of Na+ and Cl−, which are much smaller than the IL ions, enable them to occupy lattice sites of approximately the same size, thus forming very favourable lattice energies and creating a favourable situation for solid formation. This does not indicate preferential ion pairing in the ionic liquid solution, merely that the crystallized solid is thermodynamically appealing.
Seddon23 previously reported the phenomenon of immiscible ionic liquids, which indicates substantially non-ideal mixing behavior. However, each of these cases involves systems containing ions mixed (or de-mixed) together which have appreciably different sizes. These immiscible ILs form biphasic systems wherein the predominant cation and anion in each phase are matched by size – for example, when [C2C1im][C1SO3] was mixed with [C14(C6)3P][NTf2], the smaller [C2C1im]+ and [C1SO3]− ions dominated the upper phase while the larger [C14(C6)3P]+ and [NTf2]− ions dominated the lower phase. Once again, there was no indication in this experiment of preferential association within each phase, though there was preferential association between phases. These examples illustrate systems where ion association between multiple phases is dominated by entropic pressures (size matching) with no illustration of enthalpic association (ion pairing) within the IL phase(s). Mixtures of [CnC1im]Cl with [C14(C6)3P]Cl were reported as immiscible for all n < 6, despite the common anion shared between these salts. The authors also report that for all n > 1 the ΔH of mixing was negligible (between −2 and +2 kJ mol−1) while the TΔS of mixing was dominant (between −4.5 and −12 kJ mol−1),23 further indicating that mismatched cation size (entropy) is responsible for this phenomenon.
Energy of the metathesis reaction
In an ideal mixture there must be no energy change upon site exchange. In the case of these IL solutions, this implies that any ion metathesis taking place within the solution will not result in a change to the overall energy of the solution. Since Kosower's salt is present as a dilute component in these IL solutions, we assume that at most one of the anions present in the solvation shell of any cation is an iodide (all others are the anion of the ionic liquid). We also ignore interactions common to both sides (the remaining anion–cation interactions, in which only the IL anion is present), assuming that any changes on metathesis would be symmetrical. This enables us to estimate the differences in the energies of the left and right hand sides of Fig. 3 by reducing the reaction to a single metathesis (for [Py]I/[C4C1im][OTf]) (Scheme 2). | ||
| Scheme 2 | ||
The spatial arrangement of the anions around each cation allows a multitude of stable configurations. To allow a reasonable analysis within the precision of the simple description of the solvation processes as a metathesis, for [C4C1im]+ the most stable cation conformer has been selected.24 Furthermore, the two most stable ion pairs with both anions were calculated. For [Py]+ the two most stable conformers of each ion pair were calculated, ignoring the orientation of the methyl ester group. Thus, for each pair of ions there is a low energy pair {IP}low and a high energy pair, {IP}high. The energy for the metathesis reaction is therefore ambiguous.
| ΔEr = {E([Py]I) + E([C4C1im][OTf])} − {E([Py][OTf]) + E([C4C1im]I)} | (4) |
As a fixed point of reference, the most stable ion pairs were chosen for the calculation of ΔGr. Further possible energies at different levels of theory and choosing other conformers can be found in the supplementary information. However, at the highest level of theory tested by us there is no significant preference for either reactants or products of the metathesis reaction with a Gibbs free energy of −0.69 kJ mol−1, which is well within the error introduced by the simple model system.
These calculations indicate that the ion exchange energies for the [Py]I/[C4C1im][OTf] system are near zero (i.e. nearly ideal). This would mean that:
ΔGex = −RTln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kex ≈ 0 Kex ≈ 0 | (5) |
Conclusion
In stark contrast to its behaviour in molecular solvents, our experimental results show that the ionic charge-transfer salt 1-ethyl-4-(methoxycarbonyl)pyridinium iodide, Kosower's salt, does not form ion pairs in the room temperature ionic liquids studied, but rather ideal solutions. These ionic liquids are, therefore, “super-dissociating” solvents for this solute, because they completely divorce the solute cations and anions from each other. However, this does not mean that the pyridinium ions and iodide ions are never in contact, just that this contact is random. In molecular solvents, ionic species can exist as contact ion pairs, solvent-separated ion pairs or solvated free ions, but in each case the solute cation and anion require each other's proximity in order to preserve charge neutrality. Ionic liquids, conversely, solvate individual solute ions completely as the ionic liquid itself is capable of preserving charge neutrality. In this way, a salt dissolved in an ionic liquid is so dissociated as to be made into completely separate, unrelated, highly screened species. Our results are based upon observations of 1-ethyl-4-(methoxycarbonyl)pyridinium iodide in 6 different ionic liquids. These are mixtures of salts composed of similar ions, particularly in terms of relative sizes. It is possible that mixtures of more dissimilar salts would deviate from the ideal behaviour that we see here. This would be precisely analogous to the behaviour of molecular mixtures, in which structurally very similar molecules form ideal mixtures while structurally dissimilar molecules deviate from ideality.Finally, when probing the nature of ionic liquid–solute interactions using Kosower's salt, two apparently contradictory results arise. The position of the absorption maximum (Z-scale) indicates that the ionic liquids are solvents of only moderate polarity, whereas the absorptivities of the same solutions show that they are highly polar. Since these two values come from a single measurement, this difference cannot be attributed to some change in conditions or other experimental artefact. This provides an explanation of the discrepancies seen between other methods of studying ionic liquid polarity, particularly those based upon reaction kinetics7,8 and Weingärtner's dielectric spectroscopy measurements.11 Any model that involves the movement of molecules or ions necessarily implies the importance of timescales. The Z-scale measurement results from the measurement of a rapid electronic transition and is affected by a local environment that does not have the opportunity to reorganise itself on this timescale.9 This ‘freezes out’ ionic movement and the ‘snapshot’ of the ionic liquid that is obtained ostensibly appears nonpolar, as with the dielectric spectroscopy measurements. On the other hand, the absorptivity measurements are equilibrium concentration measurements wherein the longer timescale allows ion motion to dominate solvation, as with the kinetic measurements. This yields a much higher polarity. Hence, the answer to the question of how polar are ionic liquids very likely depends upon when you ask.
Methods
Computational
All calculations were performed using the Gaussian 03 software.25 Structures were optimised and frequency analysis (confirming minima and determining zero point energies and thermodynamic quantities) were carried out using the B3LYP functional.26,27 A small core ECP and the corresponding augmented polarizable double-ζ basis set have been employed for iodide.28 A 6-311+G(d,p) basis set used for all other elements.29 Zero point energy and basis set superposition error30 were calculated on this level of theory as well as MP2/6-31++G(d,p)//B3LYP/6-31++G(d,p) single point energies using Møller–Plesset perturbation theory.31Materials and reagents
1-Methylimidazole, 1-methylpyrrolidine and ethyl isonicotinate were purchased from Sigma-Aldrich and distilled from potassium hydroxide. 1-Chlorobutane was purchased from Acros Organics and distilled from phosphorus pentoxide. Lithium bis(trifluoromethylsulfonyl)imide and lithium trifluoromethanesulfonate were purchased from Solvent Innovation GmbH and used as received. All molecular solvents were purified by distillation from standard drying agents. All syntheses were performed under anaerobic conditions using standard Schlenk techniques. The preparations and spectral data of the ionic liquids have been described elsewhere.32Synthesis of 1-ethyl-4-(methoxycarbonyl)pyridinium iodide
Methyl isonicotinate (75 cm3, 635 mmol) and iodoethane (220 cm3, 2.75 mol) were heated at 40 °C for 24 h. The resulting bright orange solid was washed several times with cold acetone and EtOAc. The solid was then recrystallized from acetone to give bright orange crystals. Mp. 111.6–111.8 °C (lit. 111–112 °C);14δH (400 MHz, DMSO-d6)/ppm 9.35 and 8.52 (4H, AB quartet, 3JAB = 6.4 Hz, Py-H), 4.75 (2H, quartet, 3JH–H = 7.3 Hz, CH2CH3), 3.99 (3H, s, CO2CH3), 1.58 (3H, t, 3JH–H = 7.2 Hz, CH2CH3); δC (400 MHz, DMSO-d6)/ppm 163.1 (s,![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O2CH3), 146.55 (s, HNCH2CH3), 144.02 (s, CCO2CH3), 127.64 (s, CHCCO2CH3), 57.40 (s, NCH2CH3), 54.37 (s, CO2CH3), 16.73 (s, CH2CH3); (ESI+) MS (m/z) 166.1, [C9H12NO2]+, 100%; (ESI−) MS (m/z) 126.9, I−, 100%; 419.8, {[C9H12NO2]I2−}, 20%; Elemental analysis, found (calcd): %C = 36.86 (36.88), %H = 4.09 (4.13), %N = 4.69 (4.78).
O2CH3), 146.55 (s, HNCH2CH3), 144.02 (s, CCO2CH3), 127.64 (s, CHCCO2CH3), 57.40 (s, NCH2CH3), 54.37 (s, CO2CH3), 16.73 (s, CH2CH3); (ESI+) MS (m/z) 166.1, [C9H12NO2]+, 100%; (ESI−) MS (m/z) 126.9, I−, 100%; 419.8, {[C9H12NO2]I2−}, 20%; Elemental analysis, found (calcd): %C = 36.86 (36.88), %H = 4.09 (4.13), %N = 4.69 (4.78).
Spectroscopic measurements
Electronic spectra were obtained with a Perkin Elmer 650 UV-Vis spectrometer. Quartz cuvettes of 0.10, 0.50 and 1.00 cm pathlength were used. Z values vary with temperature,14 hence a thermostatic water circulator was used to control the sample-holder temperature to 25.0 °C.Acknowledgements
The authors are grateful for financial support from the EPSRC (MYL), the Royal Society (PAH), BASF (HN) and the European Research Council.Notes and references
- C. Reichardt and T. Welton, Solvents and solvent effects in organic chemistry, VCH Wiley, Weinheim, 4th edn, 2010 Search PubMed.
- Y. Marcus, Ion solvation, Wiley, Chichester, 1985 Search PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- C. Reichardt, Green Chem., 2005, 7, 339 RSC.
- C. Qiyuan, Z. Wenming, C. Xinmin, G. Songqing, Y. Guanqun, Z. Huifang and Y. T. Zhonglin, Thermochim. Acta, 1995, 253, 33 Search PubMed.
- (a) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS; (b) P. Wasserschied and T. Welton (ed.), Ionic liquids in synthesis, VCH Wiley, Weinheim, 2nd edn, 2007 Search PubMed.
- G. Ranieri, J. P. Hallett and T. Welton, Ind. Eng. Chem. Res., 2008, 47, 638 CrossRef CAS.
- L. Crowhurst, R. Falcone, N. L. Lancaster, V. Llopis-Mestre and T. Welton, J. Org. Chem., 2006, 71, 8847 CrossRef CAS.
- C. Chiappe, M. Malvaldi and C. S. Pomelli, Pure Appl. Chem., 2009, 81, 767 CrossRef CAS.
- (a) S. N. Baker, G. A. Baker, M. A. Kane and F. V. Bright, J. Phys. Chem. B, 2001, 105, 9663 CrossRef CAS; (b) P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS.
- (a) M.-M. Huang, Y. Jiang, P. Sasisanker, G. W. Driver and H. Weingärtner, J. Chem. Eng. Data, 2011, 56, 1494 Search PubMed; (b) H. Weingärtner, Z. Phys. Chem., 2006, 220, 1395 CrossRef CAS; (c) H. Weingärtner, A. Knocks, W. Schrader and U. Kaatze, J. Phys. Chem. A, 2001, 105, 8646 CrossRef.
- J. P. Hallett, C. L. Liotta, G. Ranieri and T. Welton, J. Org. Chem., 2009, 74, 1864 CrossRef CAS.
- R. M. Lynden-Bell, Phys. Chem. Chem. Phys., 2010, 12, 1733 RSC.
- E. M. Kosower, J. Am. Chem. Soc., 1958, 80, 3253 CrossRef CAS.
- M. Pal and S. Bagchi, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2323 RSC.
- P. Hemmes, J. N. Costanzo and F. Jordan, J. Phys. Chem., 1978, 82, 387 CrossRef CAS.
- D. A. Binder and M. M. Kreevoy, J. Phys. Chem. A, 1997, 101, 1774 CrossRef CAS.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790 RSC.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409 RSC.
- A. J. Carmichael and K. R. Seddon, J. Phys. Org. Chem., 2000, 13, 591–595 CrossRef CAS.
- C. Hardacre, J. D. Holbrey, S. E. J. McMath, D. T. Bowron and A. K. Soper, J. Chem. Phys., 2003, 118, 273 CrossRef CAS.
- M. Deetlefs, C. Hardacre, M. Nieuwenhuyzen, A. A. H. Padua, O. Sheppard and A. K. Soper, J. Phys. Chem. B, 2006, 110, 12055 CrossRef CAS.
- A. Acre, M. J. Earle, S. P. Katdare, H. Rodriguez and K. R. Seddon, Chem. Commun., 2006, 2548 RSC.
- P. A. Hunt and I. R. Gould, J. Phys. Chem. A, 2006, 110, 2269 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877 CrossRef CAS.
- (a) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS; (b) P. C. Harihara and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- (a) S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553; S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef CAS.
- (a) L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192 RSC; (b) N. L. Lancaster, P. A. Salter, T. Welton and G. B. Young, J. Org. Chem., 2002, 67, 8855 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00227a |
| This journal is © The Royal Society of Chemistry 2011 |