Catalytic enantioselective assembly of complex molecules containing embedded quaternary stereogenic centres from simple anisidine derivatives†
Rafael
Leon
,
Anup
Jawalekar
,
Thomas
Redert
and
Matthew J.
Gaunt
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, United Kingdom CB2 1EW. E-mail: mjg32@cam.ac.uk; Tel: +0044 1223 336318
First published on 18th May 2011
Abstract
Here we report an electrophile-triggered, secondary amine-catalyzed, enantioselective dearomatisation process that converts simple planar anisidine derivatives into complex tricyclic structures containing a quaternary stereogenic centre embedded within a densely functionalised molecule.
Introduction and background
Many biologically active molecules exhibit quaternary stereogenic centres at the heart of their structures.1 Accessing these molecules in an asymmetric form is not trivial;2 furthermore, the synthesis of compounds with fully substituted stereocentres that display valuable orthogonal functionality for further manipulation often requires multiple chemical transformations. Accordingly, the asymmetric synthesis of these demanding motifs has inspired many research programs,3 and the development of new catalytic enantioselective strategies that rapidly install quaternary stereogenic centres within complex molecular architectures remains a significant and important challenge to the continued advancement of chemical synthesis (eqn 1).4
Oxidative dearomatisation provides a powerful method of building complex molecules from simple starting materials;5,6 particularly useful is oxidative dearomatisation of para-substituted phenols in the presence of nucleophiles to form symmetrical cyclohexadienones. Moreover, Feringa,7a Rovis,7b You,7c and our own group7d have identified that the dearomatisation event can be coupled with an intramolecular catalytic enantioselective desymmetrisation step (Heck, organocatalytic8 Stetter, oxy-Michael and Hayashi's7eMichael addition, respectively) to form functionalised decalin-type molecules which display a quaternary stereogenic centre at the ring junction in high enantiomeric excess.
Design strategy
As part of an overarching aim towards catalytic enantioselective processes that rapidly generate products of high molecular complexity, we questioned whether we could engineer a process that would begin with an intramolecular dearomatisation event, forming a spiro-substituted cyclohexadienone, and then intercept this reactive intermediate with a desymmetrisation reaction as part of a two directional process that would ‘zip-up’ a simple arene to a complex non-racemic product (eqn 2). Here, we report that an electrophile-triggered catalytic enantioselective dearomatisation (ECED) transformation converts simple planar anisidine derivatives into complex tricyclic structures containing a quaternary stereogenic centre embedded within a densely functionalised molecule (eqn 3). The products are isolated in good yield and excellent enantiomeric excess, display intricate architecture, contain remarkably versatile orthogonal functionality and can be accessed in just three chemical operations from commercial materials. The rigid framework that surrounds the quaternary stereogenic centre projects numerous functional groups into highly defined orientations making them amenable to applications in drug discovery. Moreover, the ECED products display the complex architecture that defines a class of alkaloid natural products called the cyclindricines (eqn 4), thereby providing a potentially rapid entry into this class of molecules.9Reaction optimisation
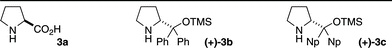
At the outset of our studies, a pivotal aspect of our design focused on identifying an intramolecular dearomatisation event triggered by an external reagent that would also install a further usable functional group in the spirocyclic cyclohexadienone product. With this in mind, we recognised that Larock's ipso-iodocyclisation of alkynyl arenes to iodine-substituted spirocyclic cyclohexadienones would provide a suitable first step for our ECED strategy.10 The electrophilic ICl activates the alkyne towards a 5-endo-dig ipso-iodocyclisation, inducing the dearomatisation that is accompanied by loss of the phenolic methyl ether group. We were able to synthesise a suitable test substrate 4a in just two steps from commercial para-iodoanisole, 4-aminobutanal diethyl acetal and phenyl propiolic acid (eqn 3) via a copper-catalyzed amination and an amide bond formation. To initially assess the reaction we chose to remove the acetal protecting group prior to dearomatisation. Treatment of 1a with ICl in anhydrous dichloromethane at −78 °C cleanly generated the iodospiro-cyclohexadienone 2a, in line with Larock's observations,10 and we immediately advanced this (without purification of 2a) to the enantioselective desymmetrising part of the ECED reaction via a secondary amine catalyzed intramolecular Michael addition.We assessed a range of catalysts (3a–c) and solvents at 0 °C and allowed reactions to reach complete consumption of starting material (Table 1); we found that derivatives of the Hayashi–Jørgensen pyrrolidine catalyst (3b–c)11 led to superior enantioselectivities when the reaction was run in dichloromethane. Further improvement to the selectivity was observed upon the addition of a benzoic acid co-catalyst, resulting in the isolation of (+)-4a in 89% yield, 92% ee, and a >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr over the two steps from aldehyde 1a (entries 8, 9). Lactam(+)-4a (absolute configuration assigned by X-ray crystallography)12 is a tricyclic structure supporting three contiguous chiral centres, comprising an embedded quaternary stereogenic centre and two tertiary chiral carbon atoms; it also displays aldehyde, enone and vinyl iodide functionality, all versatile reactive groups that can be orthogonally transformed to increase the structural complexity of the ECED products.
:1 dr over the two steps from aldehyde 1a (entries 8, 9). Lactam(+)-4a (absolute configuration assigned by X-ray crystallography)12 is a tricyclic structure supporting three contiguous chiral centres, comprising an embedded quaternary stereogenic centre and two tertiary chiral carbon atoms; it also displays aldehyde, enone and vinyl iodide functionality, all versatile reactive groups that can be orthogonally transformed to increase the structural complexity of the ECED products.

| Entry | Catalyst (mol%) | Solvent | Additive (mol%) | Temp., °C | Time, h | ee 4a, % |
|---|---|---|---|---|---|---|
| 1 | 3a(20) | MeOH | — | 0-rt | 84 | 7 |
| 2 | 3b(20) | MeOH | — | 0 | 15 | 55 |
| 3 | 3c(20) | MeOH | — | 0 | 24 | 22 |
| 4 | 3b(20) | MeCN | — | 0 | 12 | 71 |
| 5 | 3c(20) | MeCN | — | 0 | 12 | 80 |
| 6 | 3b(20) | CH2Cl2 | — | 0 | 5 | 84 |
| 7 | 3c(20) | CH2Cl2 | — | 0 | 20 | 71 |
| 8 | 3b(20) | CH2Cl2 | BzOH(20) | 0 | 14 | 92 |
| 9 | 3b(15) | CH2Cl2 | BzOH(15) | 0 | 36 | 90 |
|
||||||
Reaction scope
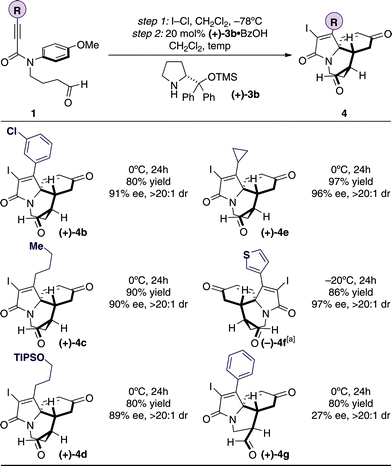
The capacity of the new transformation was next investigated by examining the scope of the alkyne substituent (Table 2); our preliminary investigations showed that aryl, heteroaryl, alkyl, cyclopropyl, and protected oxygen substituents were all accommodated by the ECED process and gave excellent yields of the tricyclic products in >90% ee and >20:1 dr using the optimised conditions 4b–f.
| a Reaction with diethylacetal derivative and catalyst(S)-3b at −20 °C. |
|---|
|
Although the ECED process also works on substrates that contain a shorter linking unit between the aldehyde and amide function to form the five-membered ring, the ee of the reaction was low in comparison to the longer chain homologue (27% ee, 4g).13 An electron rich thiophene group is not affected by the iodocyclization conditions,10 and gives the desired product (–)-4f in excellent yield and ee. We were also pleased to find that the ECED process can also be performed directly on the acetal precursors, precluding the extra step to reveal the aldehyde motif, and without compromising the yield and enantiomeric excess of the reaction (see 4f).
The ECED transformations shown in Table 2 display the electrophile and nucleophile in separate anisidine side chains, leading to a two directional closure to the tricyclic structure. Accordingly, the scope of the reaction was further extended through the successful demonstration that the aldehyde motif and electrophilic alkyne can be incorporated into the same chain 5, leading to a ‘linear’ ECED process (Scheme 1). In this system the ECED transformation forms a similar, but constitutionally different tricyclic ring system (+)-6a in excellent yield and enantiomeric excess.
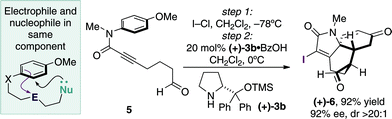 | ||
| Scheme 1 Linear ECED process. | ||
Taking advantage of the orthogonal functionality displayed by these complex quaternary centre containing molecules, we showed that the aldehyde motif in (+)-4a undergoes selective reductive amination to (+)-7a or Wittig olefination to (+)-7b, and the vinyl iodide is ideal for Pd(0)-catalyzed cross coupling (Scheme 2). We were pleased to find that selective Suzuki cross coupling with 3-pyridine boronic acid (77%, (+)-8a) and Buchwald–Hartwig amination with morpholine (86%, (+)-8b) provided representative examples to demonstrate this functionalisation idea.12 Taken together, these simple transformations provide branching points along numerous vectors that will enable the chemist to probe the chemical space around this rigid complex small molecule scaffold. Furthermore, each of these processes occur in excellent yields, do not affect the structural framework of the molecule and would be compatible with library synthesis.
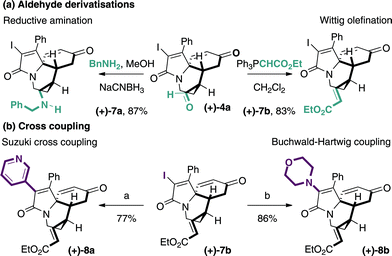 | ||
| Scheme 2 Orthogonal manipulation of functionality (a) cat. Pd2dba3, XPhos, 3-pyridine boronic acid, K3PO4, n-BuOH, 100 ˚C, (b) cat. Pd2dba3, BINAP, morpholine, Cs2CO3, PhMe, 120 ˚C. | ||
While our complexity generating transformation provides numerous opportunities to exploit the potential of post-ECED orthogonal functionalisation, we are restricted by the nature of the alkyne substituent that we need to install as part of our starting materials. To address this limitation, we speculated that a bromo-substituted alkyne motif may also be compatible with the ECED process and would accordingly result in a versatile handle for further functionalisation. Moreover, using selective cross coupling techniques it should be possible to discriminate the C–I motif and the C–Br bond, introducing different groups at each position, thereby dramatically increasing the efficacy of the ECED process (eqn 4).
Accordingly, bromo-acetal 9 was assembled in two steps from 4-iodoanisole and was successfully subjected to the ECED transformation to form the desired tricyclic structure (+)-10 in a pleasing 89% yield, >20:1 dr and 92% ee but importantly displaying the vicinal C–I and C–Br bonds (Scheme 3a). We tested the selective functionalisation strategy using iterative Pd-catalyzed cross-coupling and showed that Sonogashira coupling with phenylacetylene proceeded selectively at the C–I group in 85%12 yield to (+)-12, and the C–Br bond could subsequently be reacted through Suzuki coupling with phenyl boronic acid in 90% yield to give (+)-13, demonstrating proof of concept for the highly versatile ECED strategy (Scheme 3b).
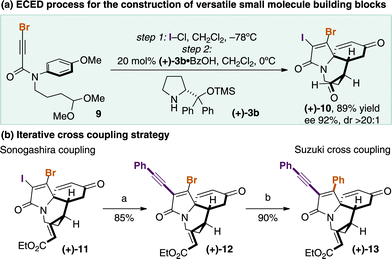 | ||
| Scheme 3 Towards the synthesis of novel complex scaffolds with selectively manipulated functionality (a) cat. PdCl2(OAc)2, CuI, phenylacetylene, Et3N, PhH, 60 ˚C; (b) cat. Pd(OAc)2, PPh3, phenylboronic acid, K2CO3, PhH-H2O, 60 ˚C. | ||
Conclusions
In summary, we have developed an electrophile-triggered catalytic enantioselective dearomatisation (ECED) transformation that converts simple anisidine derivatives into complex tricyclic structures containing a quaternary stereogenic centre embedded in a densely functionalised molecule. The efficacy of this process is reflected by the ease of which novel, highly composite and enantioenriched scaffolds can be assembled from commercial materials. Furthermore, the architecturally complex molecules display multiple stereochemical features and orthogonal functional groups that can be utilised in downstream diversification. The ECED transformation should be broadly applicable in natural product synthesis (such as the cyclindricines) and in the synthesis of novel molecules of therapeutic interest; current investigations are focused on these applications and will be reported in due course.Acknowledgements
We gratefully acknowledge the EPSRC (EP/I002065), the Marie Curie Foundation (R.L., A.J.), the DAAD (T.R.), the Royal Society and Philip & Patricia Brown (M.J.G.) for Fellowships, Novartis for funding, the EPSRC Mass Spectrometry service (University of Swansea), and Dr Nadine Gaunt Bremeyer for useful discussion and assistance with manuscript preparation.References
- E. Peterson and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11943 CrossRef CAS.
- Quaternary Stereocentres: Challenges and Solutions for Organic Synthesis; J. Christophers and A. Baro; Wiley-VCH: Weinheim, 2006 Search PubMed.
- For recent reviews on catalysis: see (a) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969 CrossRef CAS; (b) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS; (c) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS; (d) M. Hatano and K. Ishihara, Synthesis, 2008, 1647 CAS; (e) B. M. Trost and C. H. Jiang, Synthesis, 2006, 369 CrossRef CAS; (f) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (g) J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476 CrossRef CAS; (h) R. P. Wurz, Chem. Rev., 2007, 107, 5570 CrossRef CAS. For other key contributions not covered in the above reviews, see (i) F. Gao, K. P. McGrath, Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 14315 CrossRef CAS; (j) Guzzman- Martinez and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10634 CrossRef CAS and references therein; (k) M. E. Muratore, C. A. Holloway, A. W. Pilling, R. I. Storer, G. Trevitt and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 10796 CrossRef CAS.
- For selected examples of representative catalysis modes in natural product synthesis: see (a) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488 CrossRef CAS; (b) J. M. Enquist and B. M. Stoltz, Nature, 2008, 453, 1228 CrossRef; (c) Y. Saga, R. Motoki, S. Makino, Y. Shimizu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS; (d) P. Jakubec, D. M. Cockfield and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 16632 CrossRef CAS.
- For a recent review of oxidative dearomatisation in natural product synthesis, see (a) L. Pouysegu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235 CrossRef CAS . See also; (b) M. A. Ciufolini, N. A. Braun, S. Canesi, M. Ousmer, J. Chang and D. Chai, Synthesis, 2007, 3759 CrossRef CAS.
- For examples of asymmetric oxidative dearomatisation in natural product synthesis, see: (a) L. H. Mejorado and T. R. R. Pettus, J. Am. Chem. Soc., 2006, 128, 15625 CrossRef CAS; (b) S. Dong, J. Zhu and J. A. Porco Jr, J. Am. Chem. Soc., 2008, 130, 2738 CrossRef CAS; (c) J. Qi, A. B. Beeler, Q. Zhang and J. A. Porco Jr, J. Am. Chem. Soc., 2010, 132, 13642 Search PubMed.
- (a) R. Imbos, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 184 CrossRef CAS; (b) Q. Liu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 2552 CrossRef CAS; (c) Q. Gu, Z.-Q. Rong, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2010, 132, 4056 CrossRef CAS; (d) N. T. Vo, R. D. M. Pace, F. O'Hara and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 404 CrossRef CAS; (e) Y. Hayashi, H. Gotoh, T. Tamura, H. Yamaguchi, R. Masui and M. Shoji, J. Am. Chem. Soc., 2005, 127, 16028 CrossRef CAS.
- For general review on organocatalysis, see: (a) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS ; For enamine catalysis, see; (b) S. Mukherjee, J. Woon Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS.
- For a recent example of the synthesis of cyclindrincine C see: T. J. Donohoe, P. M. Brian, G. C. Hargaden and T. J. C. O'Riordan, Tetrahedron, 2010, 66, 6411 Search PubMed.
- X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2005, 127, 12230 CrossRef CAS.
- (a) Y. Hayashi, M. Gotoh, T. Hayashi and M. Shioji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS; (b) M. Marigo, T. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., 2005, 44, 804 Search PubMed.
- See supporting information for details†.
- While we are not certain of the reason for the low selectivity observed in 4g, it is possible that a under the reaction conditions a non catalysed pathway is competing with the catalytic enamine mediated reaction, rather than due to low selectivity in enamine Michael addition.
Footnote |
| † Electronic supplementary information (ESI) available: experimental data for all new compounds and procedures. The structure of 4a has been deposited in the Cambridge Crystallographic Data Centre (CCDC reference number 808822). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00218j |
| This journal is © The Royal Society of Chemistry 2011 |