Steady state and transient kinetics in crystalline solids: the photochemistry of nanocrystalline 1,1,3-triphenyl-3-hydroxy-2-indanone†
Gregory
Kuzmanich
a,
Jiadan
Xue
c,
José-Carlos
Netto-Ferreira
*b,
J. C.
Scaiano
b,
Matthew
Platz
*c and
Miguel A.
Garcia-Garibay
*a
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095-1559, USA. E-mail: mgg@chem.ucla.edu; Tel: +1 (310) 825 3159
bDepartment of Chemistry, University of Ottawa, Ottawa, ON K1N 6N5, Canada. E-mail: jcnetto@ufrrj.br; Tel: +1-(613) 562-5633
cDepartment of Chemistry, Ohio State University, Colombus, OH 43210, USA. E-mail: platz@chemistry.ohio-state.edu; Tel: +1 (614) 292-0401
First published on 19th May 2011
Abstract
Electronic excitation of crystalline 1,3,3-triphenyl-1-hydroxy-2-indanone results in the exclusive formation of hydroxy-benzocyclobutane while irradiation in solution leads to formation of the isomeric photoenols, which subsequently tautomerize. Using nanocrystals suspended in water we were able to use transmission pump–probe methods to detect the reactive intermediates involved in the solid state. Transients assigned to the 1,4-biradical (triplet enol) formed by adiabatic photodecarbonylation were detected. It was found that product formation occurs from analogous transients, both in solution and in the solid state. While enols revert to ketones in time scales that range from a few hundred nanoseconds to tens of microseconds, benzocyclobutanol remains kinetically trapped in the crystal lattice, but undergoes a thermal ring opening when dissolved.
Introduction
Photochemical reactions in crystals have potential for applications in materials science1 and in solvent-free, green chemical synthesis. While the rigid environment of crystals prevents many photochemical reactions, it is well known that those with limited molecular motion can be strongly favored and their selectivity controlled to remarkable extents.2 Unfortunately, the development and application of reactions in crystals have been slow, in part due to the lack of mechanistic insights analogous to those available for reactions in solution. While there has been promising advances in time-resolved single crystal X-ray crystallography,3 reaction mechanisms in crystals have been generally limited to those that can be inferred from product analysis.4 The use of optical methods to detect and determine the kinetics of excited states and reactive intermediates would be ideal. However, studies in large single crystals and bulk powders are hampered by their high optical densities and strong light scattering, though the use of diffuse reflectance methods is sometimes helpful.5 To address these limitations in a very simple manner, it has been shown that nanocrystals in the 150–300 nm range, with sizes that are smaller than the wavelength of excitation, are amenable to pump–probe optical and magnetic resonance methods when suspended in water.6 In this communication, we report the first transient analysis and kinetic characterization of Norrish type I decarbonylation of 2-indanone 1 in solution and as a nanocrystalline suspension.Based on our experience with the photoinduced decarbonylation of crystalline ketones, we decided to use 1,3,3-triphenyl-1-hydroxy-2-indanone 1 as a test system. Previous work7,8,9 has shown that photochemical decarbonylation of several 2-indanones gives different products in solution and in the solid state, making the phase of the reaction critical. In this article we present the steady state photochemistry and transient absorption studies of 2-indanone 1 (Scheme 1) in solution, in dry solids, and as a nanocrystalline suspension in water. As reported previously, irradiation in solution resulted in the formation of photoenols EE-2 and ZZ-2, which transform thermally into ketone 4 within a few hundred nanoseconds or a few microseconds, respectively. In contrast, reactions in crystals gave exclusively the benzocyclobutanol 3, though the latter ring opens and converts into ketone 4 very rapidly upon dissolution. As shown in Scheme 1, pump–probe studies in the picosecond and nanosecond regimes suggest that the reaction proceeds by the same excited state transients in solution and in the solid state.
 | ||
| Scheme 1 Photolysis of 1 as a solvated molecule, or in a nanocrystalline suspension, intersystem crosses to the triplet state before reacting to give the common intermediate 331BR-2BR-2. In solution, photoenols 2 are formed, but in the nanocrystals, benzocyclobutane 3 is formed exclusively. | ||
Results and discussion
The synthesis of 1 has been described previously9c,10 and gram quantities can be obtained in four steps from 2,3-diphenyl 1-indanone in an overall yield of 44% (SI†). Compound 1 was characterized via1H and 13C NMR, FTIR, and UV-Vis, all of which were identical to those reported earlier (SI†). Indanone 1 is a white crystalline solid with a melting point of 158 °C. A X-ray structure determined in single crystals grown from ethanol was solved in the space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules per unit cell.‡ As illustrated in Fig. 1, the two molecules form a self-complementary centrosymmetric dimer with hydrogen bonds between the α-hydroxyl group of one molecule and the carbonyl of the other. The α,α′,α′ phenyl rings have edge-to-face contacts with phenyl rings of adjacent molecules (Figure S20†).
with two molecules per unit cell.‡ As illustrated in Fig. 1, the two molecules form a self-complementary centrosymmetric dimer with hydrogen bonds between the α-hydroxyl group of one molecule and the carbonyl of the other. The α,α′,α′ phenyl rings have edge-to-face contacts with phenyl rings of adjacent molecules (Figure S20†).
 | ||
| Fig. 1 Single-crystal X-ray structure of 1 showing the intermolecular hydrogen bonding between adjacent molecules to form a centrosymmetric complex. | ||
Nanocrystalline suspensions were prepared by rapidly injecting a concentrated acetone solution of 1 into rapidly vortexing water (SI†).11Dynamic light scattering showed that the average particle size is ca. 208 nm, and a scanning electron microscopy (SEM) image on a silicon wafer demonstrates particle sizes between ca. 75 and 200 nm (Fig. 2). X-Ray powder diffraction patterns of the bulk and nanocrystalline material are very similar to each other as well as with the diffraction pattern calculated from the single crystal X-ray structure, suggesting a single polymorph (Figure S12†).
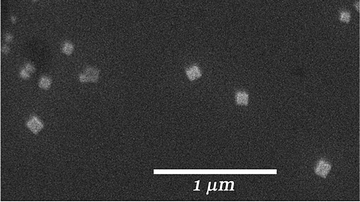 | ||
| Fig. 2 SEM image of isolated crystalline nanoparticles of 1 of ca. 75–200 nm in size. | ||
Direct irradiation of 1 in acetonitrile, cyclohexane, or benzene at 312 nm resulted in a 100% yield of the o-benzyl benzophenone 4 at low conversion (ca. 20%) (SI†).
Irradiation of bulk powders or nanocrystalline suspensions of 1 also resulted in 4 when detected after dissolution. However, in situ analysis by solid state FTIR of crystals exposed to UV light, later shown to have reacted to 23% conversion, revealed peaks consistent with the exclusive formation of benzocyclobutane 3 (Fig. 3). A broad hydrogen-bonded OH stretch from ca. 3100–3500 cm−1 in crystalline 1 narrows upon irradiation indicating the formation of the free tertiary hydroxyl of benzocyclobutane 3. No peaks characteristic of ketone 4 are observed in the reacted solid. However, the spectrum obtained after dissolution and subsequent recrystallization of irradiated 1 shows a new band at 1661 cm−1, which is assigned to the diarylcarbonyl found in 4. These observations indicate that cyclobutanol 3 transforms into ketone 4 upon dissolution (Fig. 3).
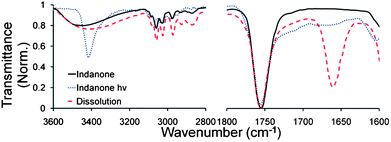 | ||
Fig. 3
IR spectra of 1 as a bulk solid before (—) and after irradiation (⋯) indicating no formation of benzophenone 4. Upon dissolving the crystal in DCM (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), the benzocyclobutane thermally rearranges to give benzophenone 4. ), the benzocyclobutane thermally rearranges to give benzophenone 4. | ||
Optically dense nanocrystalline suspensions of photoactive organic compounds in water also provide a simple and effective way to measure the quantum yields of reaction.12 Using the decarbonylation of dicumyl ketone (Φ-CO = 0.18)13 as a chemical actinometer, the quantum yield of decarbonylation of 1 and formation of 3 in the solid state is ΦNC = 0.10. While benzophenone 4 is detected upon GC analysis, it is assumed that benzocyclobutane 3 converts to benzophenone 4 thermally and quantitatively. Using valerophenone in Ar-purged benzene (ΦBen. = 0.33, λEx = 312 nm),14 it was determined 1 has a Φ−CO = 0.29 in the same solvent.
Given that the quantum yields of reactions in nanocrystals and in benzene were reasonably high (0.1 and 0.3, respectively), transient absorption spectroscopy was first attempted on the nanosecond timescale. Previous work has shown that photodecarbonylation of 1 in methanol or trifluoroethanol results in formation of photoenols ZZ-2 and EE-2 within the 10 ns laser pulse.15 In excellent agreement, measurements carried out in acetonitrile, λEx = 255 nm, led to one transient with λmax at 330 nm with a secondary maximum at 460 nm (Figure S15†) that is consistent with the transients previously assigned for the overlapping photoenols ZZ-2 and EE-2.15 Lifetimes of 300 ns and 30 μs for ZZ-2 and EE-2, respectively, are also in good agreement (Figure S16†). In contrast to excitation in acetonitrile, laser flash photolysis of 1 as a nanocrystalline suspension in water, λEx = 255 nm, produced no detectable transients on the nanosecond timescale. However, analysis of the suspension after laser irradiation indicated that product formation had occurred up to ca. 10%, implying that either the transients are too weak to observe between 260 and 600 nm on the nanosecond timescale, or that they have lifetimes that are shorter than 10 ns. In order to answer this question we investigated the formation of transients on faster time scales in cyclohexane solutions (CHX) and in nanocrystalline suspensions (NC).
Gratifyingly, using 50 fs excitation pulses at 255 nm with a time window of up 3 ns we detected relatively strong signals between 400–600 nm, both in solution and in the solid state (Fig. 4). While transients in solution and in nanocrystals showed some similarities and only small spectral changes, three decay components could be resolved in each case. Measurements in cyclohexane showed the formation of a broad transient immediately after the pulse with λmax = 460 nm which decays in ca. 2 ps (kdec = 8 × 1011s−1). A second component shifts the spectrum to a λmax = 470 nm and proceeds with a rate of kdec = 1.3 × 1010s−1. The latter transient decays with a rate of kdec = 7.1 × 108 s−1 to a new transient λmax = 525 nm, which is persistent for the duration of the experiment (3 ns). Measurements in nanocrystals gave relatively similar results. The spectrum present after the laser pulse contains a transient species with λmax = 460 nm which decays biexponentially (kdec = 5 × 1011 and 9.8 × 109s−1), the third component shifts to λmax = 505 nm (kdec = 2.2 × 108 s−1), and the latter transient shifts to λmax = 520 nm (>3 ns). While definitive assignments are not possible given the limited literature data available and the broad nature of the spectra, the time scales involved in these processes are consistent with events that precede the formation of the ground state photoenol and the cyclobutanol, in each of the two reaction media. As indicated in Scheme 1, the transients that precede the formation of 3 and 4 are the singlet (111*) excited state and the biradicals that result from α-cleavage (111BR-1BR-1 and 331BR-1BR-1), and from decarbonylation (331BR-2BR-2).
 | ||
| Fig. 4 Transient absorption spectra of 1 as a nanocrystalline suspension showing the prompt formation of the acetophenone ketyl-like transient 111BR-1BR-1 (460 nm) at early times, and the benzophenone ketyl-like transient 331BR-2BR-2 (505 nm) at later times. | ||
A sequence that involves the generally accepted sequence: 111* → 331* → 331BR-1BR-1 → 331BR-2BR-2 → product, seems unlikely. To the best of our knowledge, there are no absorptions of singlet excited states (S1 → Sn) for aliphatic ketone in this region of the spectrum, and the absorption of excited ketone triplets (T1 → Tn) occurs at a λmax ≈ 300 (e.g.acetone), which is far from the transients observed in this study.§ In contrast, it is well known that chromophores associated with the ketyl, diphenylmethyl, and trityl radicals embedded in the structures of 1BR-1BR-1 and 1BR-2BR-2 absorb in this spectral region. It is also known that the molar absorptivity of ketyl radicals is ca. 3 orders of magnitude greater than those of diphenyl methyl and trityl radicals,16 suggesting that the subnanosecond transients observed are all associated with transitions centered on the ketyl radical fragments. We propose that the most likely species are those associated with the transients highlighted in Scheme 2: an acetophenone ketyl-like absorption with λmax ≈ 450 nm and a benzophenone ketyl-like transient with λmax ≈ 540 nm. The trityl radical has been previously reported to have a λmax at 335 nm and a secondary maximum at 440 nm, neither of which were detected.17 Recent calculations reported by Zipse suggest that the ketyl radical is 4.2 kJ mol−1 more stable than the trityl radical formed by cleavage of the other σ-bond.18
 | ||
| Scheme 2 Transients observed after femtosecond excitation have λmax between 400–550 nm. The most likely chromophores are the embedded acetophenone and benzophenone moieties in 331BR-1BR-1 and 331BR-2BR-2. | ||
Assuming that none of the observed transients are associated with the singlet or triplet excited states of 1,¶ but that they are associated instead with ketyl radical transients, a possible kinetic scenario is the rapid formation of a vibrationally excited 1BR-1BR-1 from 111* which undergoes relaxation (1.4 ps in CHX and 2.0 ps in NC) to vibrationally relaxed 11BR-1.19 Consistent with this assignment, it is well known that cyclic aliphatic ketones with radical-stabilizing α-substituents may undergo α-cleavage reactions from the singlet excited state with rates that are greater than those for intersystem crossing to the triplet.20 Tsentalovich et al. have shown that acyl–alkyl biradicals undergo ISC with rates kISC = 2.20 x109s−1,21 and based on this we suggest that the next transient produced in our studies may be 33BR-1, which forms in competition with reversible bond formation back to the ground state ketone. This assignment indicates that intersystem crossing of the acyl-alkyl biradical from 1 occurs with rates of kISC = 1.3 × 1010s−1 and 9.8 × 109s−1 in solution and in crystals, respectively, which are in excellent agreement with the literature report.21
The longest-lived transients may be assigned to 33BR-2, which has a spectrum that shifts to a λmax ≈ 520 nm in the solid and λmax ≈ 525 nm in solution, in agreement with the previously reported spectra of 33BR-2 in methanol.15a The suggested decarbonylation of the acyl radical intermediate, with rate constants of k-CO = 7.1 × 108s−1 and k-CO = 2.2 × 108s−1 in solution and in crystals, occurs with a similar rate as that for the decarbonylation of the diphenylmethyl acyl radical (Ph2MeCCO˙), k-CO = 1.5 × 108 s−1.22 The lifetime of 33BR-2 should be limited by intersystem crossing and formation of benzocyclobutanol 3 in the solid state, which apparently occurs before 10 ns (the time resolution of the ns experiments). In solution, the formation of the photoenols ZZ-4 and EE-4 also occur in the nanosecond timescale.15
Because suspension and solution photoreactions proceed through the same intermediates, steric effects in the crystal play a key role in the bifurcation leading to the final product. Indanone 1 crystallizes in a closely packed manner with close contacts on all three α-phenyl rings. While formation of benzocyclobutane 3 requires minimal motion of the phenyl rings for cyclization to occur, formation of either photoenol, ZZ-2 or EE-2, requires rotation about both α-carbons to achieve the nearly planar conformation of the photoenol. Space filling models indicate that rotation about these bonds to form planar 2 would be severely hindered.
Conclusions
Steady state irradiation of 1 as a nanocrystalline suspension and in solution results in two structurally distinct photoproducts: benzocyclobutanol 3 and o-benzyl benzophenone 4, respectively. We have shown through the use of steady state and transmission laser flash photolysis on solution and nanocrystalline suspensions that these products derive from a common reactive intermediate, 331BR-1BR-1. Consistent with previous reports, this common intermediate forms the photoenols ZZ-2 and EE-2 in solution. However, the crystal packing of 1 inhibits formation of the photoenols but does allow bond formation to yield benzocyclobutanol 3. Femtosecond spectroscopy suggests that this reaction proceeds via α-cleavage from the singlet state, followed by ISC of the acyl–alky biradical to the triplet. Decarbonylation of the acyl radical in solution and nanocrystals occurs before ring closure (crystalline) or enolization (in solution).Acknowledgements
This research was supported the NSF grants DMR0937243 and CHE0844455. G. K. acknowledges NSF IGERT MCTP grant DGE0114443.Notes and references
- (a) M. L. Calvo and P. Cheven, ed. A. T. Friberg and R. Daendliker, SPIE Publications, Bellingham, WA, 2008, p. 285; (b) K. Milam, G. O'Malley, N. Kim, G.D. and T. Kyu, J. Phys. Chem. B, 2010, 114, 7791 Search PubMed; (c) S. Das, S. Varghese and N. S. S. Kuman, Langmuir, 2010, 26, 1598 CrossRef CAS; (d) J. Ge, H. Lee, L. He, J. Kim, Z. Lu, H. Kim, J. Goebl, S. Kuran and Y. Yen, J. Am. Chem. Soc., 2009, 131, 15687 CrossRef CAS; (e) M. Irie and M. Morimoto, Pure Appl. Chem., 2009, 81, 1655 CrossRef CAS.
- (a) S. M. Mandel, P. N. Singh, S. Muthukrishnan, M. Chang, J. A. Krause and A. D. Gudmundsdóttir, Org. Lett., 2006, 8, 4207 Search PubMed; (b) J. Luo, H. Ihmels, H.-J. Deiseoth and M. Schlosser, Can. J. Chem., 2009, 87, 619 Search PubMed; (c) H. E. Zimmerman and G. A. Sereda, J. Org. Chem., 2002, 68, 283 Search PubMed; (d) J. Sankaranarayanan, L. N. Bort, S. M. Mandel, P. Chen, J. A. Krause, E. E. Brooks, P. Tsang and A. D. Gudmundsdóttir, Org. Lett., 2008, 10, 937 Search PubMed.
- (a) J. Davaasambuu, G. Busse and T.S., J. Phys. Chem. A, 2006, 110, 3261 CrossRef CAS; (b) M. T. Miller, B. O. Bachmann, C. A. Townsend and A. C. Rosenzweig, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14752 CrossRef CAS; (c) J. Hallmann, W. Morgenroth, C. Paulmann, J. Davaasambuu, Q. Kong, M. Wulff and S. Techert, J. Am. Chem. Soc., 2009, 131, 15018 Search PubMed.
- (a) P. Naumov, K. Sakurai, M. Tanaka and H. Hara, J. Phys. Chem. B, 2007, 111, 10373 Search PubMed; (b) A. Natarajan, C. K. Tsai, S. I. Khan, P. McCarren, K. N. Houk and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2007, 129, 9846 CrossRef CAS.
- (a) F. Wilkinson and C. J. Willsher, Appl. Spectrosc., 1984, 38, 897 Search PubMed; (b) F. Wilkinson, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2073 RSC.
- (a) N. V. Lebedeva, V. F. Tarasov, M. J. E. Resendiz, M. A. Garcia-Garibay, R. C. White and M. D. E. Forbes, J. Am. Chem. Soc., 2010, 132, 82 CrossRef CAS; (b) S. Simoncelli, G. Kuzmanich, M. N. Gard and M. A. Garcia-Garibay, J. Phys. Org. Chem., 2010, 23, 376 CAS; (c) K. K. Chin, A. Natarajan, L. M. Campos, E. Johansson, H. Shepherd and M. A. Garcia-Garibay, Chem. Commun., 2007,(41), 4266 RSC.
- (a) L. M. Campos, C. J. Mortko and M. A. Garcia-Garibay, Mol. Cryst. Liq. Cryst., 2006, 456, 15 Search PubMed; (b) M. A. Garcia-Garibay and L. M. Campos, ed. W. Horspool and F. Lenci, CRC Press, Boca Raton, FL, 2003; (c) L. M. Campos, D. Ng, Z. Yang, H. Dang, H. L. Martinez and M. A. Garcia-Garibay, J. Org. Chem., 2002, 67, 3749 CrossRef CAS.
- (a) G. Quinkert, J. Palmowski, H. P. Lorenz, W. W. Wiersdorff and M. Finke, Angew. Chem., Int. Ed. Engl., 1971, 10, 198 Search PubMed; (b) G. Quinkert, Photochem. Photobiol., 1968, 7, 783 Search PubMed.
- (a) D. Leinweber and H. Butenschön, Tetrahedron Lett., 1997, 38, 6385 Search PubMed; (b) D. Ng, Z. Yang and M. A. Garcia-Garibay, Tetrahedron Lett., 2002, 43, 7063 CrossRef CAS; (c) T. Paul, M. A. Hassan, H.-G. Korth, R. Sustmann and D. V. Avila, J. Org. Chem., 1996, 61, 6835 CrossRef CAS.
- (a) C. F. Koelsch, J. Org. Chem., 1938, 3, 456 Search PubMed; (b) C. F. Koelsch and C. D. Le-Claire, J. Org. Chem., 1941, 6, 516 Search PubMed.
- H. Kasai, H. S. Nalwa, H. Oikawa, S. Okada, H. Matsuda, N. Minami, A. Kuakuta, K. Ono, A. Mukoh and H. Nakanishi, Jpn. J. Appl. Phys., 1992, 31, 1132.
- (a) G. Kuzmanich, A. Natarajan, K. K. Chin, M. Veerman, C. J. Mortko and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2008, 130, 1140 CrossRef CAS; (b) G. Kuzmanich, M. N. Gard and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2009, 131, 11606 CrossRef CAS.
- M. Veerman, M. J. E. Resendiz and M. A. Garcia-Garibay, Org. Lett., 2006, 8, 2615 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Marcel Dekker, Inc., New York, 1993 Search PubMed.
- (a) J. C. Netto-Ferreira, V. Wintgens and J. C. Scaiano, J. Braz. Chem. Soc., 1997, 8, 427 Search PubMed; (b) J. C. Netto-Ferreira, V. Wintgens and J. C. Scaiano, Can. J. Chem., 1994, 72, 1565 CAS; (c) J. C. Scaiano, V. Wintgens and J. C. Netto-Ferreira, Pure Appl. Chem., 1990, 62, 1557 CAS.
- (a) C. Chatgilialoglu, in Handbook of Organic Photochemistry, ed. J. C. Scaiano, CRC Press, Boca Raton, Florida, 1989, pp. 3–11 Search PubMed; (b) J. C. Scaiano, A. Martin, G. P. A. Yap and K. U. Ingold, Org. Lett., 2000, 2, 899–901 CrossRef CAS.
- J. L. Faria and S. Steenken, J. Am. Chem. Soc., 1990, 112, 1277 CrossRef CAS.
- H. Zipse, Top. Curr. Chem., 2006, 263, 163 CAS.
- A similar scenario was observed in the femotosecond decarbonylation studies of acetone excited to S2: E. W.-G. Diau, C. Kötting, T. I. Sølling and A. H. Zewail, ChemPhysChem, 2002, 3, 57 Search PubMed.
- (a) N. C. Yang, E. D. Feit, M. H. Hui, N. J. Turro and J. C. Dalton, J. Am. Chem. Soc., 1970, 92, 6974 CrossRef CAS; (b) J. C. Dalton, D. M. Pond, D. S. Weiss, F. D. Lewis and N. J. Turro, J. Am. Chem. Soc., 1970, 92, 2564 Search PubMed; (c) D. Weiss, Organic Photochemistry, Padwa, A., Ed., Marcel Deker: New York, 1981, Vol. 5, pp 347 Search PubMed.
- Y. P. Tsentalovich, O. B. Morozova, N. I. Avdievich, G. S. Anachenko, A. V. Yurkovskaya, J. D. Ball and M. D. E. Forbes, J. Phys. Chem. A, 1997, 101, 8809 Search PubMed.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and photochemical procedures and the .cif file for indanone 1. CCDC reference number 818702. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00184a |
| ‡ The diffraction data from a crystal of indanone 1 were solved in the triclinic space groupP (formula C27H20O2, a = 8.514, b = 8.71, c = 13.415, α = 92.829°, β = 103.590°, γ = 102.480°, Vol/Å3 = 968.8, Z = 2). |
| § Even the acetophenone triplet has a λmax = 330 nm with a very weak absorption in the 400 nm range. |
| ¶ The use of triplet quenchers showed no significant effect on the reaction of 1 suggesting an upper lifetime of ca. 0.1–1 ns for a potential triplet excited state precursor. |
| This journal is © The Royal Society of Chemistry 2011 |