Quasi-biomimetic ring contraction promoted by a cysteine-based nucleophile: Total synthesis of Sch-642305, some analogs and their putative anti-HIV activities†
Alpay
Dermenci
a,
Philipp S.
Selig
a,
Robert A.
Domaoal
b,
Krasimir A.
Spasov
b,
Karen S.
Anderson
*b and
Scott J.
Miller
*a
aDepartment of Chemistry, Yale University, 225 Prospect Street, New Haven, CT 06520-8107, USA. E-mail: scott.miller@yale.edu; Fax: (+1) 203-496-4900
bDepartment of Pharmacology, Yale University School of Medicine, 333 Cedar Street, SHM B350B, New Haven, CT 06520, USA. E-mail: karen.anderson@yale.edu; Fax: (+1) 203-785-7670
First published on 20th June 2011
Abstract
Cysteine plays a number of important functional and structural roles in nature, often in the realm of catalysis. Herein, we present an example of a cysteine-promoted Rauhut-Currier reaction for a potentially biomimetic synthesis of Sch-642305 and related analogs. In this key step of the synthesis we discuss interesting new discoveries and the importance of substrate-catalyst recognition, as well as cysteine's structural features. Also, we investigate the activity of Sch-642305 and four analogs in HIV-infected T-cells.
Introduction
Natural products remain inspirational entities for study of chemical issues, biosynthetic hypotheses and as platforms for the development of biologically active compounds. We report herein our studies of the natural product Sch-642305 (Fig. 1),1,2 which have ventured into each of these scientific venues.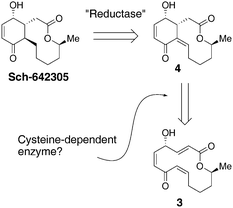 | ||
| Fig. 1 A plausible biosynthetic route to Sch-642305. | ||
Our interest in Sch-642305 began in the context of our studies of the cysteine-catalyzed Rauhut-Currier reaction. This process allows catalytic enantioselective ring-forming reactions of the type shown in eqn (1) (1 → 2, 95% ee).3 After development of this reaction, we were stimulated to ask whether or not cysteine-catalyzed ring-forming processes were documented in the context of natural product biosynthesis. For example, might Sch-642305 be assembled biologically through the action of a cysteine-dependent enzyme that might mediate the ring contraction of 3 to deliver 4? Reductases then could tailor the final formation of the natural product. While a number of stellar laboratory-based natural product total syntheses have relied upon phosphine-induced Rauhut-Currier reactions,4 including a suggestion that a biological Rauhut-Currier might take place in the context of spinosyn biosynthesis,5 to our knowledge cysteine-dependent enzymes that mediate such processes are not yet well described.6 The catalysis of such biosynthetic steps by alternative biologically available thiols (glutathione, co-enzyme A, etc.) is also possible. Yet, the plausible existence of such enzymes within the unannotated proteome may now be emerging as a significant question in biosynthetic circles.7 We thus wondered if studies of cysteine-dependent ring contractions could lead to some sense of plausibility for related natural product biosynthesis hypotheses.
 | (1) |
The screening of natural products' biological activities also leads to provocative questions about natural products' endemic functions, and of course their possible alternative functions as therapeutics. In this sense, Sch-642305 has been examined, and some possible biological functions have been ascribed, including a possible connection to the HIV-1 Tat protein,8 an essential component for viral replication.9 Very often, first-pass in vitro screening studies do not allow for follow-up in vivo analysis. Moreover, supply problems and a lack of structurally related analogs can lead to a lack of resolution on the potential of many natural product hits. We have been fortunate in our studies of Sch-642305 to see our chemical hypotheses culminate in the synthesis and study of a number of analogs that have led to a higher resolution sense of this natural product's mode of action in this biological context.
Results and discussion
Our assessment of the possible cysteine-catalyzed ring contraction required the synthesis of an appropriate macrocyclic precursor. For this goal, we designated macrocycle 5 as the appropriate substrate (Fig. 2). This compound allows us to pose additional questions about the prowess of cysteine-based catalysts for the ring contraction step: Is catalyst chirality essential for stereocontrol during the conversion of 5 to 6? By way of analogy, would an enzyme be essential during the putative ring contraction in order to control diastereoselectivity during the bond-forming event? Or, on the other hand, is the stereochemical outcome pre-programmed into the stereostructure of the substrate?10 The use of chiral catalysts to control diastereoselectivity is a highly challenging arena for the field of asymmetric catalysis.11 The relevance of this issue for stereocontrolled biosynthesis is also one that is perhaps not systematically understood. Moreover, could catalytic hydrogenation conditions be found that would deliver not only 7 from 6, but also alternative diastereomers that might shed light on the structure–activity relationship of Sch-642305? | ||
| Fig. 2 Retrosynthetic analysis that allows for possible condition-dependent synthesis of stereoisomeric analogs. | ||
Synthesis of our Rauhut-Currier substrate commenced with alkene 8 (Scheme 1). Epoxidation furnished 9,12 which was resolved using the Jacobsen hydrolytic kinetic resolution13 to achieve 10 in 45% yield and >97.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 e.r. Opening of the epoxide to allylic alcohol 11 (87% yield),14 followed by cross metathesis,15 TBDPS protection, and TMSOK-mediated hydrolysis provided fragment 12 in 74% yield (3 steps).
:2.5 e.r. Opening of the epoxide to allylic alcohol 11 (87% yield),14 followed by cross metathesis,15 TBDPS protection, and TMSOK-mediated hydrolysis provided fragment 12 in 74% yield (3 steps).
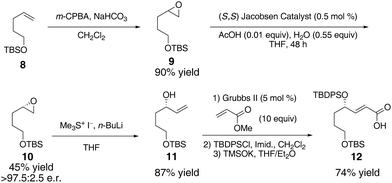 | ||
| Scheme 1 | ||
The second portion of the substrate was synthesized starting with TBS protection and ozonolysis of known alcohol 1316 to obtain 14 in 95% yield over two steps (Scheme 2). Takai olefination17 with a CrCl2/CHI3 combination followed by TBS removal gave alcohol 15 in 64% yield. The two fragments 12 and 15, which we were able to synthesize on a multi-gram scale, were brought together using DCC and DMAP. Deprotection and oxidation of the coupled product furnished the Nozaki–Hiyama–Kishi (NHK) substrate 16 in 64% yield over three steps.18 A macrocyclization under NHK conditions (CrCl2/NiCl2 (99:1) in DMSO) followed by an oxidation of the resulting allylic alcohol provided the 14-membered macrocycle (5) in 59% yield over two steps.
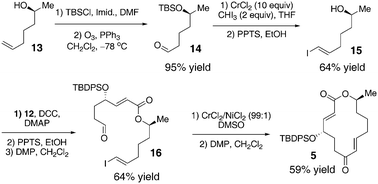 | ||
| Scheme 2 | ||
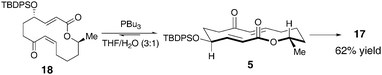
We were thus in a position to assess the macrocyclic substrate 5 for its propensity to engage in ring contractionvia the Rauhut-Currier reaction. A productive reaction entails addition of the catalyst to the substrate, proper conformational alignment for a transannular Michael addition, and successful elimination of the catalyst. Using a well-precedented catalyst for ring contractions of this type (PBu3, 1 equiv),4 we observed the formation of one major product (>30:1, 17) in 80% yield (Scheme 3). This result may suggest that upon catalyst addition the substrate adopts a single low energy conformation that allows transannular Michael addition upon one face of the enoate. Of note, deprotection of 17 under buffered TBAF conditions provided 6, the structure of which was confirmed by X-ray crystallography. Also, the olefin formed upon catalyst elimination was thus verified to be in the E configuration.
 | ||
| Scheme 3 | ||
With these encouraging results in hand, we wondered what the outcome would be by altering the olefin geometry of the enone in the substrate. When substrate 1819 was subjected to the phosphine-catalyzed conditions the same product was obtained in slightly lower yield (65%), but with the same selectivity (>30:1, eqn (2)). During the course of the reaction the catalyst transforms the substrate from 18 to the lower energy isomer 5 (monitored by TLC) and continues to product 17. From these results we realized that a great barrier must be overcome to reverse the diastereoselectivity of the substrate.
 | (2) |
We then turned our attention to the study of a potentially biomimetic cysteine-based catalytic ring contraction. As a starting point we subjected substrate 5 to the conditions shown in eqn (1) and were disappointed to find little to no reactivity. Nonetheless, after extensive screening and optimization, the (R)-enantiomer of the cysteine-based catalyst (19) and the conditions in Fig. 3 provided 17 in 66% yield. Increasing the amount of base to 9 equiv and diluting the reaction mixture (0.05 M to 0.01 M)20 proved to be the most crucial factors for obtaining higher conversion. On the other hand, when the enantiomeric catalyst 20 was used, no reaction took place over the course of the same time frame. The dependence of the outcome of the reaction – indeed the notion that the chirality of the catalyst is critical for the success or failure of the ring contraction – speaks to the sensitivity of these reactions to issues of “matching” and “mismatching” in accord with the principles of double diastereodifferentiation.21 They also prompted an experiment with an achiral catalyst. Indeed, when the reaction is attempted with isobutyl thiol (21) as the putative catalyst, a Rauhut-Currier reaction does not result. Instead, in accordance with the pioneering observations of Murphy,22 alternative products are observed wherein elimination of the catalyst has not occurred. Thiol conjugate addition product 22 and the ring-contracted, thiol-containing adduct 23 are produced in a ∼1:1 ratio, in a combined yield of 81%. These results are consistent with the idea that catalyst regeneration is promoted by chelation effects involving the additional functionality associated with cysteine-based catalysts. While these observations by no means necessitate the involvement of enzymes in reactions that could be speculated to occur biosynthetically, they do underscore the need for additional functionality when thiol and thiolate-based catalysts are used for these processes. The dependence of the outcome on the stereochemical identity of the catalyst also speaks to the degree of transition state organization that is required.
 | ||
| Fig. 3 Cysteine-catalyzed Rauhut-Currier ring contraction. | ||
The chirality-based dependence of the reaction stimulated an additional question about the degree to which the chiral catalysts would influence the stereochemical outcome of an analogous Rauhut-Currier reaction devoid of pre-existing stereogenic centers. For example, would substrate 24 be amenable to enantioselective ring contraction under the influence of catalyst 19? Macrocycle 24 was thus prepared by methods analogous to those employed for the synthesis of 5.23 Indeed, as shown in Table 1, an enantioselective Rauhut-Currier ring contraction was possible under the influence of catalyst 19. Under carefully defined conditions, product 25 could be isolated in as high as 65% ee, although yields of the reaction were modest (25%).

With these observations in hand, we addressed the completion of the synthesis of Sch-642305. As in our proposed biomimetic hypothesis, a directed hydrogenation using [Rh(COD)dppb]+BF4− under high pressure H2 (1000 psi) for 24 h afforded the desired reduction product (26) in 81% yield and 12:1 d.r. (Scheme 4). Presumably, the hydroxyl is a directing group in delivering the catalyst from the bottom face of the olefin.24 Protection of the alcohol followed by Saegusa oxidation gave us compound 27 in 44% yield over two steps. Finally, deprotection of the TBS-protected alcohol led to Sch-642305 (69% yield),25 which we were able to verify by X-ray crystallography.
 | ||
| Scheme 4 | ||
Our synthetic strategy provides an opportunity for divergent syntheses of Sch-642305 analogs. Thus, we sought to synthesize the epimeric form of Sch-642305. This required reducing the double bond of the Rauhut-Currier product from the opposite face of the olefin. Subjecting intermediate 17 to hydrogenation conditions shown in Scheme 5 afforded the desired diastereomer (28) in 69% yield as a 5:1 mixture of separable isomers. Saegusa oxidation followed by deprotection produced epi-Sch-642305 in 63% yield over two steps.
 | ||
| Scheme 5 | ||
The set of analogs we obtained enabled an assessment of structure–activity relationships for Sch-642305 in the context of its putative activity against HIV-1 as well as general cytotoxicity (Fig. 4). Compounds were tested against the wild-type IIIB strain of HIV-1 in MT-2 human T-cells at a viral multiplicity of infection of 0.1.26 Efavirenz was a compound used as a positive control having potent antiviral activity in the low nanomolar range with a lack of general cytotoxicity. We determined the EC50, which is the antiviral potency of the compound and is represented by the closed circles on the curves. This is the concentration at which 50% of the cells are protected from cell death due to viral killing. The IC50 is the general cytotoxicity of the compound and is represented by the open circles on the curves. It is the concentration at which 50% of the cells are killed by the compound in the absence of virus. We found that Sch-642305 showed some activity against the virus that was above background but below 50% protection. It was also toxic to the MT-2 cells at an IC50 of 0.75 μM. The epimeric form of Sch-642305 had a 6-fold reduction in toxicity compared to Sch-642305 with an IC50 of 4.5 μM but did not appear to show any antiviral activity. The analogs, 26, 29, and 6, further improved the toxicity profile with IC50 values greater than 100 μM. Interestingly, these results support the previously reported in vitro anti-HIV potential of Sch-642305, which appears to be absent with our structurally related analogs. Several interesting features of structure activity relationships for Sch-642305 and related analogs emerge from this analysis. First, only the natural product, Sch-642305, showed both a hint of antiviral activity and cellular toxicity highlighting the importance of α,β-cyclohexenone ring unsaturation and stereochemistry. Second, the presence of an α,β-unsaturation within the cyclohexenone appears to be a determinant of cytotoxicity since only Sch-642305 and its epi form show cellular toxicity. If the unsaturated bond is absent (as for 26 and 29) or exocyclic (6), little or no toxicity is observed at concentrations up to 100 μM.
 | ||
| Fig. 4 Testing of compounds toward HIV-1 in MT-2 human T-cells. | ||
Taken together, the attendant cytotoxicity and putative antiviral activity of the natural product in in vivo studies suggests that extrapolation to therapeutic potential will not be straightforward. However, the observed cytotoxicity of Sch-642305 is also largely specific to the natural product structure, and not the analogs, and these studies could well be of interest in future biological studies of this scaffold.
Conclusions
In summary, we have developed synthetic schemes that enable the total synthesis of Sch-642305, and several structurally related analogs. The synthetic strategy was stimulated by questions about a possible role for cysteine- or other thiol-based catalysts in a putative biosynthetic pathway. While these possibilities remain unproven from the biological perspective, chemical plausibility was demonstrated with the use of a single amino acid. Furthermore, these experiments showcase the importance of the resident functionality of the peptide backbone in the cysteine-promoted Rauhut-Currier reaction. Also, these case studies led us to investigate asymmetric variants of the ring contraction, which ultimately resulted in an intriguing example of an enantioselective transannular Rauhut-Currier reaction. We hope that our findings can offer some insight for the presence of the Rauhut-Currier in biological systems.These synthetic studies allowed preparation of an expanded set of Sch-642305-like compounds. In vivo assessment of their biological activities confirmed a possible role for inhibitory behavior of Sch-642305 in the HIV life cycle. However, only a hint of activity was observed before parallel cytotoxicity led to cell death in these studies. Notably, the slight activity that was observed was unique to the natural product, while the alternative structural analogs did not demonstrate these effects.
Acknowledgements
We are grateful to the National Science Foundation (CHE-0848224) and National Institute of Health (GM49551) to K.S.A. for support. We would also like to thank Dr. Carrie E. Aroyan for preliminary work involving synthesis of the Rauhut-Currier substrates. Dr. Christopher Incarvito is gratefully acknowledged for assistance with X-ray crystallographic data. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, is also greatly appreciated: MT-2 cells, catalog no. 237, from Douglas Richman. P.S.S. is grateful to the German Academic Exchange Service DAAD for a postdoctoral fellowship.Notes and references
- M. Chu, R. Mierzwa, L. Xu, L. He, J. Terracciano, M. Patel, V. Gullo, T. Black, W. Zhao, T.-M. Chan and A. T. McPhail, J. Nat. Prod., 2003, 66, 1527 CrossRef CAS.
- For previous syntheses please see: (a) G. Mehta and H. M. Shinde, Chem. Commun., 2005, 3703 RSC; (b) B. B. Snider and J. Zhou, Org. Lett., 2006, 8, 1283 Search PubMed; (c) K. Ishigami, R. Katsuta and H. Watanabe, Tetrahedron, 2006, 62, 2224 Search PubMed; (d) E. M. Wilson and D. Trauner, Org. Lett., 2007, 9, 1327 Search PubMed; (e) H. Fujioka, Y. Ohba, K. Nakahara, M. Takatsuji, K. Murai, M. Ito and Y. Kita, Org. Lett., 2007, 9, 5605 CrossRef CAS; (f) J. Garcia-Fortanet, M. Carda and J. A. Marco, Tetrahedron, 2007, 63, 12131 Search PubMed.
- C. E. Aroyan and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 256 CrossRef.
- (a) L.-C. Wang, A. L. Luis, K. Agapiou, H.-Y. Jang and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 2402 CrossRef CAS; (b) S. A. Frank, D. J. Mergott and W. R. Roush, J. Am. Chem. Soc., 2002, 124, 2404 CrossRef CAS; (c) K. Agapiou and M. Krische, Org. Lett., 2003, 5, 1737 CrossRef; (d) J. L. Methot and W. R. Roush, Org. Lett., 2003, 5, 4223 CrossRef CAS.
- (a) S. A. Frank and W. R. Roush, J. Org. Chem., 2002, 67, 4316 CrossRef CAS; (b) D. J. Mergott, S. A. Frank and W. R. Roush, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11955 CrossRef CAS; (c) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (d) S. M. Winbush, D. J. Mergott and W. R. Roush, J. Org. Chem., 2008, 73, 1818 CrossRef CAS.
- For an alternative mechanistic possibility, based on a putative “enolase,” see: H. J. Kim, R. Pongdee, Q. Wu, L. Hong and H.-W. Liu, J. Am. Chem. Soc., 2007, 129, 14582 Search PubMed.
- (a) H. J. Kim, M. W. Ruszczycky, S.-h. Choi and H.-w. Liu, Nature, 2011, 473, 109 Search PubMed; (b) Private communication from Prof. Kendall Houk, University of California at Los Angeles.
- H. Jayasuriya, D. L. Zink, J. D. Polishook, G. F. Bills, A. W. Dombrowski, O. Genilloud, F. F. Pelaez, L. Herranz, D. Quamina, R. B. Lingham, R. Danzeizen, P. L. Graham, J. E. Tomassini and S. B. Singh, Chem. Biodiversity, 2005, 2, 112 CrossRef CAS.
- J. Karn, J. Mol. Biol., 1999, 293, 235 CrossRef CAS.
- E. J. Sorensen, Bioorg. Med. Chem., 2003, 11, 3225 Search PubMed.
- E. P. Balskus and E. N. Jacobsen, Science, 2007, 317, 1736 CrossRef CAS.
- K. C. Nicolaou, R. Gorduru, Y.-P. Sun, B. Banerji and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2007, 46, 5896 CrossRef CAS.
- (a) M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936 CrossRef CAS; (b) S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS.
- D. F. Taber and Z. Zhang, J. Org. Chem., 2006, 71, 926 Search PubMed.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS.
- D. J. Dixon, S. V. Ley and E. W. Tate, J. Chem. Soc., Perkin Trans. 1, 2000, 2385 RSC.
- M. Tortosa, N. A. Yakelis and W. E. Roush, J. Am. Chem. Soc., 2008, 130, 2722 CrossRef CAS.
- (a) Y. Okude, S. Hirano, T. Hiyama and H. Nozaki, J. Am. Chem. Soc., 1977, 99, 3179 CrossRef CAS; (b) L. Venkatraman, C. E. Salomon, D. H. Sherman and R. A. Fecik, J. Org. Chem., 2006, 71, 9853 CrossRef CAS.
- Substrate 18 was synthesized using a similar route to 5, where the Z vinyl iodide of 15 was synthesized. Please see Electronic Supporting Information for procedure (Scheme SI-1).†.
- Using freshly distilled MeCN over CaH2 was critical as any possible amounts of hydrolyzed MeCN (acetamide) could retard reaction rates.
- S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed. Engl., 1985, 24, 1 CrossRef.
- (a) F. Dinon, E. Richards, P. J. Murphy, D. E. Hibbs, M. B. Hursthouse and K. M. Malik, Tetrahedron Lett., 1997, 40, 3279 Search PubMed; (b) P. M. Brown, N. Käppel and P. J. Murphy, Tetrahedron Lett., 2002, 43, 8707 CrossRef CAS; (c) P. M. Brown, N. Käppel, P. J. Murphy, S. J. Coles and M. B. Hursthouse, Tetrahedron, 2007, 63, 1100 CrossRef CAS.
- Please see Electronic Supporting Information (Scheme SI-2). †.
- J. M. Brown, Angew. Chem., Int. Ed. Engl., 1987, 26, 190 CrossRef.
- Previously developed conditions (ref. 2d) using TBAF/AcOH led to substrate epimerization.
- (a) T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS; (b) C. Pannecouque, D. Daelemans and E. De Clerq, Nat. Protoc., 2008, 3, 427 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, HPLC traces, biological assay, spectroscopic and crystallographic data for all new compounds. CCDC reference numbers 820817–820821. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00221j |
| This journal is © The Royal Society of Chemistry 2011 |