Structural variety in iridate oxides and hydroxides from hydrothermal synthesis†
Kripasindhu
Sardar
a,
Janet
Fisher
b,
David
Thompsett
b,
Martin R.
Lees
c,
Guy J.
Clarkson
a,
Jeremy
Sloan
c,
Reza J.
Kashtiban
c and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
bJohnson Matthey Technology Centre, Sonning Common, Reading, RG4 9NH, UK
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 23rd May 2011
Abstract
We report the results of an exploratory synthetic study of iridium-containing materials using hydrothermal methods from simple metal salts. Three alkali-earth iridium hydroxides are isolated as phase-pure samples and their structures examined by single-crystal or powder diffraction methods: each contains Ir(IV)-centred octahedra, isolated from each other and sharing bridging hydroxides or fluoride with alkali-earth (Ca, Sr or Ba) centres. One of these hydroxides, Ca2IrF(OH)6.OH, possesses a unique open structure, consisting of a positively-charged framework that has one-dimensional channels in which infinite chains of hydrogen-bonded hydroxide anions are encapsulated. The addition of hydrogen peroxide or sodium peroxide to otherwise identical hydrothermal reactions yields dense oxide materials in which iridium is found in an oxidation state between +4 and +5: the novel oxide Na0.8Sr2.2Ir3O10.1 has a KSbO3-type structure with an iridium oxidation state of +5, while the new pyrochlore (Na0.27Ca0.59)2Ir2O6·0.66H2O contains iridium with an average oxidation state close to +4.5. Our results illustrate the utility for hydrothermal synthesis in the discovery of novel complex structures that may be inaccessible using conventional high-temperature synthesis, with control of the metal oxidation state possible with judicious choice of reagents.
Introduction
In the synthesis of novel solid-state materials that have extended structures, an important strategy is the exploration of a wide variety of chemical reaction conditions.1 Jansen and co-workers have used the analogy of the exploration of an energy landscape2 where metastable phases may be isolated that would usually be inaccesible in conventional synthetic solid-state chemistry because high temperatures and pressures would typically be used to induce reaction between solid precursors. This may allow the discovery of new phases but also control of the crystal form to optimise properties; particularly important for electronic, optical or catalysis applications. The use of methods such as co-precipitation, sol–gel and molten-salt reaction media are indeed now well-established means of removing the high energy input needed in solid-state synthesis to prepare, in particular, oxide solids.3 The use of a solvent brings further scope for control in oxide synthesis, where tuning of not only chemical composition and atomic-scale structure but also crystal morphology may be possible; this is particularly appropriate for the preparation of nanostructured materials.4 In this respect, hydrothermal (more generally solvothermal) synthesis, well-known for the preparation of open-framework structures such as zeolites, zeotypes and metal–organic frameworks,5 has some considerable application in the preparation of dense oxides6 and a number of anhydrous, ceramic materials have already been isolated as fine powders in single-step reactions without post-synthesis annealing; these may have nanostructure, and in some cases unusual polymorphs not seen at high temperature and pressures can also be stabilised.7,8 Until now there has been a particular focus on the hydrothermal synthesis of mixed-metal oxides of the first transition-metal series, including titanates, manganites and ferrites for their ferroelectric and magnetic properties;8 in contrast, there are few reports of the hydrothermal chemistry of heavier, late transition-metals of the precious-metal groups, where the only examples have focussed on ruthenium oxides.9 Herein we demonstrate how mild hydrothermal chemistry provides a versatile introduction to the chemistry of complex, iridium-containing hydroxides and oxides: the oxides of this metal are of topical interest for their applications in electrocatalysis.10Results and discussion
The hydrothermal reaction11 at 195 °C between iridium(III) chloride and strontium nitrate in aqueous sodium hydroxide solution yields a phase-pure sample of a novel hydroxide Sr2Ir(OH)8, whose structure was solved and refined using single-crystal X-ray diffraction, Fig. 1a. The material is an analogue of a known tin compound Sr2Sn(OH)8,12 and its structure is constructed from two unique Sr atoms, each with dodecahedral coordination, and an iridium centre with octahedral coordination. The strontium centres are linked to each other by pairs of bridging hydroxide ions and to the iridium centres by further bridging hydroxides that triply bridge two strontiums and one iridium in total. The network formed has voids running along [001] into which the hydrogen atoms of the hydroxide ions project. Upon heating in air above 300 °C, in situ thermodiffractometry reveals that Sr2Ir(OH)8 decomposes to iridium metal along with strontium carbonate rather than a mixed oxide phase: this is in contrast to the tin analogue that yields a mixture of SrSnO3 and Sr(OH)2.12 The oxidation state of iridium in Sr2Ir(OH)8 was confirmed as +4 using bond valence sums,13 which gave a value of +4.07, and also by the use of Ir LIII-edge X-ray absorption near edge spectroscopy (XANES),14Fig. 2a, where a distinctive double feature is seen in the second derivative spectrum. This double feature has been explained previously as being characteristic of isolated octahedral {IrO6} units (i.e. not sharing oxygens with neighbouring iridium centres), and has been ascribed as arising from well-resolved 2p-3d(t2g) and 2p-3d(eg) transitions at the absorption edge.15,16 The XANES data also confirm the lower oxidation state of Ir(III) in the iridium chloride precursor (an amorphous material by powder X-ray diffraction) and thus shows how the use of aqueous solutions of nitrate precursors and sodium hydroxide provides oxidising reaction conditions for the isolation of Ir(IV) materials.![Views of the crystal structures of (a) Sr2Ir(OH)8 (viewed along [001]) (b) Ca2IrF(OH)6·OH (viewed along [001]) and (c) BaIr(OH)6·H2O showing part of one layer found in the ab plane. In each case the iridium-centred octahedra are coloured orange, in (b) oxygens of extra-framework hydroxides are shown as red spheres and in (c) the oxygen atoms of crystal water molecules are shown as red spheres.](/image/article/2011/SC/c1sc00192b/c1sc00192b-f1.gif) | ||
| Fig. 1 Views of the crystal structures of (a) Sr2Ir(OH)8 (viewed along [001]) (b) Ca2IrF(OH)6·OH (viewed along [001]) and (c) BaIr(OH)6·H2O showing part of one layer found in the ab plane. In each case the iridium-centred octahedra are coloured orange, in (b) oxygens of extra-framework hydroxides are shown as red spheres and in (c) the oxygen atoms of crystal water molecules are shown as red spheres. | ||
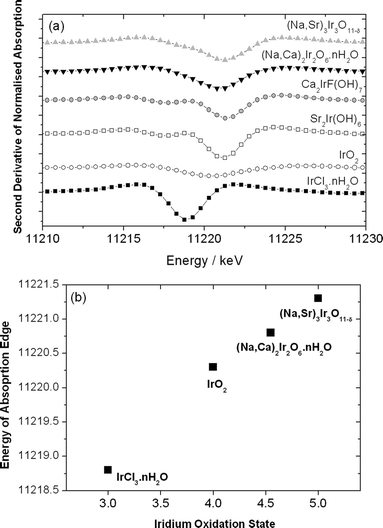 | ||
| Fig. 2 Ir LIII-edge XANES of new phases and reference materials: (a) Plots of the second derivative spectra and (b) the edge position determined by the minimum in the second derivative spectrum and corresponding to 2p-3d transitions plotted versusoxidation state determined from the chemical composition of selected compounds. | ||
Similar reactions with calcium nitrate as a precursor do not yield a crystalline calcium-iridium hydroxide unless NaF is included. Initially we tested this reagent as a mineraliser, to aid solubilisation of reagents and hence enhance crystallisation, but the material formed as single crystals actually contains stoichiometric amounts of fluorine. Ca2IrF(OH)6·OH possesses a unique open structure, Fig. 1b, in which iridium(IV) is coordinated by an octahedron of six hydroxides that in turn bridge to the calcium ions. The eight coordination of calcium is completed by two fluoride ions forming one edge of a distorted dodecahedron. The fluoride ions themselves sit at the centre of a regular tetrahedron of four calciums, analogous to the environment seen in the archetypal ionic solid CaF2. This generates an open network that possesses positive charge, in turn balanced by hydroxide ions that lie on high symmetry sites at the centres of cylindrical channels that run along [001]. These ‘extra framework’ hydroxide ions are connected to each other by hydrogen bonds (oxygen–oxygen distance = 2.83 Å) to form infinite chains of –OH–OH–, in a similar fashion to that seen in the mineral hydroxyapatite.17 In the calcium-iridium hydroxide material, however, these ions are tightly bound: attempts at their removal by ion-exchange reactions in concentrated aqueous solids of halides, nitratesetc. had no effect on the composition, or indeed the crystallinity of the material. The lack of crystal water is confirmed by thermogravimetric analysis and IR spectroscopy (Supporting Information†) and thermal treatment shows the collapse of the material above 300 °C with continued heating to 800 °C yielding a mixture of oxides that includes Ca2IrO4, CaIrO3 and IrO2. Bond valence sums confirm the oxidation state of +4 for iridium in Ca2IrF(OH)6·OH, while XANES analysis confirms this and is also consistent with the presence of isolated, non-interacting {IrO6} centres in the solid, Fig. 2a.
When using barium reagents, analogous hydrothermal reactions yield the phase BaIr(OH)6·H2O, which is a structural analogue of the known phase BaPt(OH)6·H2O18 (see Supporting Information†). This phase also contains {IrIVO6} centres, isolated from each other and linked with nine-coordinated barium centres to give a complex layered motif separated by the crystal water, Fig. 1c in which OH groups project into the layers where crystal water is hydrogen bonded. In contrast to the strontium and calcium hydroxides, thermal decomposition of BaIr(OH)6·H2O results in a single-phase oxide product: 2H-BaIrO3 (Supporting Information†).
The addition of hydrogen peroxide to the hydrothermal reactions produces distinctively different materials to the hydroxides so far described. In the case of the strontium-iridium system, the novel oxide Na0.8Sr2.2Ir3O10.1 is formed as a phase-pure powder consisting of submicron-sized crystallites, Fig. 3b. This is a new example of an iridate that has the KSbO3+δ type structure, as proved by refinement against powder neutron diffraction data,19Fig. 3a. The presence of sodium was also confirmed by bulk analysis for metals. Previous examples of iridates with this structure type include Ba0.5IrO3 and Sr0.5IrO3, both of which are A-site deficient and have body-centred variants of the parent structure,20 and the related phase Ba2Ir3O9 that was prepared under high pressure/temperature hydrothermal conditions (150 MPa and 650 °C).21 Na0.8Sr2.2Ir3O10.1 has primitive cubic symmetry (proved by indexing the laboratory powder XRD), consistent with the stoichiometric occupancy of metal sites within the structure, but with also some additional oxide ions within the channels formed by the connected {IrO6} network, Fig. 3a. Based on the refined composition from the neutron diffraction data, the oxidation state of iridium is close to +5. This is supported by bond valence sums: although the only available parameters are for Ir(IV)13 and these give a valence of +4.6, the average Ir–O bond distance of 1.97 Å is similar to that seen in established Ir(V) oxides, such as in Ba2YIrO6 (1.96 Å)16 and La2.5K1.5IrO7 (1.97 Å).22Ir LIII-edge XANES of the new phase show an edge shift of ∼1 eV from IrO2, Fig. 2, a shift associated with oxidation state +5.15,16 In addition, the second derivative XANES spectrum shows a broad single feature, as seen also in IrO2: this has been ascribed as being due to the eg and t2g valence levels not being resolved and characteristic of the presence of interacting, connected {IrO6} units,15,16 as expected in the KSbO3-type structure.
![Characterisation data for Na0.8Sr2.2Ir3O10.1 (a) final profile refinement of powder neutron diffraction data (with view of structure along [111] inset) (b) SEM image of crystallites.](/image/article/2011/SC/c1sc00192b/c1sc00192b-f3.gif) | ||
| Fig. 3 Characterisation data for Na0.8Sr2.2Ir3O10.1 (a) final profile refinement of powder neutron diffraction data (with view of structure along [111] inset) (b) SEM image of crystallites. | ||
In the case of the calcium-iridium system, with the addition of sodium peroxide a new cubic pyrochlore material is formed as a polycrystalline material under hydrothermal conditions. Considering thermogravimetric analysis, charge-balancing considerations, and a refinement of the structure against the powder neutron diffraction data (Fig. 4a) this material is formulated as (Na0.27Ca0.59)2Ir2O6·nH2O (n = 0.66). This material contains iridium in an oxidation state only slightly more than +4.5, and its Ir LIII-edge XANES spectrum is consistent, Fig. 2, with an edge position between that of IrO2 and the new strontium phase described above. Other iridium-containing pyrochlores that have been reported include those of both Ir(IV) and Ir(V) such as Ln2Ir2O7 (Ln =lanthanide),23Cd2Ir2O720 and Ca2Ir2O7,20 as well as mixed-valent materials, Pb2-xCaxIr2O7-y,24 all prepared by the annealing of oxide precursors at elevated temperatures. TEM of the calcium-sodium iridate pyrochlore, Fig. 4b, shows highly crystalline particles and the modulation of electron density seen by HRTEM along [110] is consistent with the metal deficient A-site where the presence of sodium as well as calcium, gives columns of low electron density contrasting with the high electron density of the B site. Upon annealing the pyrochlore, which was attempted in order to make conductivity measurements, we found that heating in air even at 400 °C results in a collapse to give significant amounts of IrO2, while heating at 800 °C in air results in the formation of the known phase Ca2IrO4 along with iridium metal. In contrast to the case of strontium and calcium, in the barium-iridium system we find no crystalline material is formed when peroxide is added to similar hydrothermal reactions, under the moderate temperature reaction conditions we have used.
![(Na0.27Ca0.59)2Ir2O6·0.66H2O (a) final profile refinement of powder neutron diffraction data (with view of structure along [110] inset) and (b)–(d) TEM analysis of the material, showing an HRTEM image in (b) that is indexed as the [110] direction according to the SAED pattern in (d). A magnified region of (b) is shown in (c) with a superimposed simulation from the refined neutron diffraction structure.](/image/article/2011/SC/c1sc00192b/c1sc00192b-f4.gif) | ||
| Fig. 4 (Na0.27Ca0.59)2Ir2O6·0.66H2O (a) final profile refinement of powder neutron diffraction data (with view of structure along [110] inset) and (b)–(d) TEM analysis of the material, showing an HRTEM image in (b) that is indexed as the [110] direction according to the SAED pattern in (d). A magnified region of (b) is shown in (c) with a superimposed simulation from the refined neutron diffraction structure. | ||
A study of the magnetic properties of the five new iridium-containing materials was made as shown in Fig. 5 and 6. In the case of the three hydroxides, Ca2IrF(OH)6·OH, Sr2Ir(OH)8 and BaIr(OH)6·H2O, cooperative ordering is seen at low temperature with the first, Fig. 5b, showing a ferromagnetic component with a Curie temperature of ∼1.8 K, with a weak hysteresis with applied field, while the strontium and barium hydroxides show evidence for antiferromagnetic ordering with Néel temperatures of ∼3 K, with no hysteresis seen with applied field at low temperatures, Fig. 5a and 5c. The signs of the Weiss constants from Curie–Weiss fits, confirm the ordering behaviours (Supporting Information†). The low temperatures of these transitions are not surprising given that the materials contain isolated {IrO6} octahedra. In contrast, the two oxides remain paramagnetic down to 1.8 K. The magnetic response of these materials is modified by the octahedral crystalline electric field environment of the Ir ions. The low value of the effective magnetic moment on the Ir ions and the almost temperature-independent susceptibility seen at higher temperature Fig. 6, can be understood using the theory developed by Kotani,25 which considers the effects of spin–orbit coupling on the magnetic moment of a transition metal ion placed in an octahedral crystal field environment.
 | ||
| Fig. 5 Magnetic susceptibility data at low temperature for: (a) Sr2Ir(OH)8 (b) Ca2IrF(OH)6·OH and (c) BaIr(OH)6·H2O. Inset in each case shows the magnetisation as a function of applied field at various temperatures. ZFCW = zero field cooled warming and FCC = field cooled cooling. | ||
 | ||
| Fig. 6 Magnetic susceptibility versus temperature data for (a) Na0.8Sr2.2Ir3O10.1 and (b) (Na0.27Ca0.59)2Ir2O6·0.66H2O. In both cases the anomaly at 50–100 K is due to the condensation of paramagnetic oxygen on the fine powders and this signal is greatly reduced upon warming at 400 K under helium prior to measurement. The insets show the magnetisation with applied field at 2 K. | ||
Conclusions
The use of peroxide in the hydrothermal synthesis of oxide materials provides a convenient oxidant in the synthesis of precious-metal oxides: we find relatively high oxidation states of iridium in KSbO3-type and pyrochlore materials, dense oxides with directly linked iridium centres, instead of hydroxides that contain isolated octahedral Ir(IV). It has been noted previously that in order to prepare oxides of iridium in oxidation states greater than +5, forcing reaction conditions are needed, such as high oxygen pressure,15,16 or molten hydroxide fluxes,26 so it is not surprising that the materials prepared under the much milder conditions we have investigated contain iridium in an oxidation state no higher than +5. As well as providing a means of controlling oxidation state in hydrothermal synthesis, the peroxide also appears to plays a role in the selective formation of dense oxides in preference to open structured hydroxides. Our results thus provide an important illustration of the power of mild hydrothermal conditions in the isolation of novel, condensed crystalline materials with fine control over the composition and structural connectivity of the resulting phases. Mild conditions are employed and with the judicious choice of solution reagents, a variety of structure types may be isolated in one-step reactions with no need for post-synthesis annealing; indeed each of the materials we have prepared is metastable with respect to phase separation upon heating. It is apparent from our work that the hydrothermal chemistry of other precious metals may be worthy of study.Acknowledgements
We thank the EPSRC (UK) for funding this work (EP/F012721/1) and for providing access to the TEM instruments in Oxford Materials under the Materials Equipment Access scheme (EP/F01919X/1). We thank the STFC (UK) for provision of beamtime at Diamond Light Source and at the ISIS Neutron Source and we are grateful to Dr S. Ramos and Dr R. I. Smith, respectively, for their assistance at the central facilities. Some of the equipment used in materials characterisation at the University of Warwick was obtained through the Science City Advanced Materials project “Creating and Characterising Next Generation Advanced Materials” with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). Johnson Matthey plc kindly loaned precious metal salts.Notes and references
- C. N. R. Rao and J. Gopalakrishnan, Acc. Chem. Res., 1987, 20, 228 CrossRef CAS.
- M. Jansen, Angew. Chem., Int. Ed., 2002, 41, 3747; M. Jansen, K. Doll and J. C. Schon, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 518 CrossRef.
- J. Rouxel and M. Tournoux, Solid State Ionics, 1996, 84, 141 CrossRef CAS; J. Gopalakrishnan, Chem. Mater., 1995, 7, 1265 CrossRef CAS.
- G. R. Patzke and Y. Zhou, Angew. Chem., Int. Ed., 2011, 50, 826 Search PubMed.
- R. M. Barrer, Hydrothermal Chemistry of Zeolites, Academic Press, London, 1982 Search PubMed; A. K. Cheetham, G. Férey and T. Loiseau, Angew. Chem., Int. Ed., 1999, 38, 3268 Search PubMed; L. R. MacGillivray, Metal–Organic Frameworks Design and Application, John Wiley and Sons, Hoboken, 2010 CrossRef CAS.
- K. Byrappa and M. Yoshimura, Handbook of Hydrothermal Technology, Noyes Publications, Park Ridge, New Jersey USA, 2001 Search PubMed.
- G. Demazeau, J. Mater. Chem., 1999, 9, 15 RSC.
- R. E. Riman, W. L. Suchanek and M. M. Lencka, Ann. Chim., 2002, 27, 15 CAS; D. R. Modeshia and R. I. Walton, Chem. Soc. Rev., 2010, 39, 4303 RSC.
- H. S. Horowitz, J. M. Longo and J. T. Lewandowski, Mater. Res. Bull., 1981, 16, 489 CrossRef CAS; C.-C. Wang and C.-C. Hu, Electrochim. Acta, 2005, 50, 2573 Search PubMed; R. J. Darton, S. S. Turner, J. Sloan, M. R. Lees and R. I. Walton, Cryst. Growth Des., 2010, 10, 3819 Search PubMed.
- A. M. Kannan, A. K. Shukla and S. Sathyanarayana, J. Electroanal. Chem., 1990, 281, 339 Search PubMed; M. V. ten Kortenaar, J. F. Vente, D. J. W. Ijdo, S. Müller and R. Kötz, J. Power Sources, 1995, 56, 51 CrossRef CAS; T. Nakagawa, N. S. Bjorge and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 15578 CrossRef CAS.
- Hydrothermal synthesis was undertaken using IrCl3·5H2O as an iridium source in Teflon-lined, stainless-steel autoclaves and reaction temperatures of less than 250 °C with typical reaction times of a few days. Solid products were isolated by filtration and dried in air at 80 °C before further study. Full synthetic details are supplied in Supporting Information†. All materials were phase pure, both from visual inspection of samples and also from the results of later analysis.
- M. M. Wu, X. L. Li, G. P. Shen, J. Li, R. R. Xu and D. M. Proserpio, J. Solid State Chem., 2000, 151, 56 Search PubMed.
- A. S. Wills, VaList, 2010, Search PubMedProgram available from http://www.ccp14.ac.uk.
- Ir LIII-edge XANES spectra were recorded in transmission mode using B18 of the Diamond Light Source UK from samples of iridates diluted in polyethylene powder and pressed into self supporting pellets. Data were recorded simultaneously from a reference Ir foil placed between the transmission and a third ion chamber.
- J. H. Choy, D. K. Kim, G. Demazeau and D. Y. Jung, J. Phys. Chem., 1994, 98, 6258 Search PubMed.
- J. H. Choy, D. K. Kim, S. H. Hwang, G. Demazeau and D. Y. Jung, J. Am. Chem. Soc., 1995, 117, 8557 CrossRef CAS.
- O. Hochrein, R. Kniep and D. Zahn, Chem. Mater., 2005, 17, 1978 CrossRef CAS.
- G. Bandel, C. Platte and M. Trömel, Z. Anorg. Allg. Chem., 1981, 477, 178 Search PubMed.
- Powder neutron diffraction measurements were made using the POLARIS diffractometer at ISIS, the UK spallation neutron source, from samples contained within thin-walled, cylindrical vanadium cans at room temperature.
- A. W. Sleight, Mater. Res. Bull., 1974, 9, 1177 CrossRef CAS.
- Y. Kawamura and H. Sato, J. Alloys Compd., 2004, 383, 209 Search PubMed.
- S. J. Mugavero, M. D. Smith and H. C. zur Loye, J. Solid State Chem., 2005, 178, 3176 Search PubMed.
- B. J. Yang and Y. B. Kim, Phys. Rev. B, 2010, 82 Search PubMed.
- H. Sakai, H. Ohno, N. Oba, M. Kato and K. Yoshimura, Phys. B, 2003, 329, 1038 Search PubMed.
- M. Kotani, J. Phys. Soc. Jpn., 1949, 4, 293 CAS; T. Sakamoto, Y. Doi and Y. Hinatsu, J. Solid State Chem., 2006, 179, 2595 Search PubMed; M. Wakeshima, D. Harada and Y. Hinatsu, J. Alloys Compd., 1999, 287, 130 CrossRef CAS.
- S. J. Mugavero, M. D. Smith, W. S. Yoon and H. C. zur Loye, Angew. Chem., Int. Ed., 2009, 48, 215 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 819218–819219. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00192b |
| This journal is © The Royal Society of Chemistry 2011 |