Ferrocene-amino acid macrocycles as hydrazone-based receptors for anions†
Sophie R.
Beeren
and
Jeremy K. M.
Sanders
*
University Chemical Laboratory, University of Cambridge, Lensfield Rd, Cambridge, CB2 1EW, United Kingdom. E-mail: jkms@cam.ac.uk; Fax: +44(0)1223 336017; Tel: +44(0)1223 336411
First published on 19th May 2011
Abstract
We report the synthesis of a family of new macrocyclic hydrazone-based anion receptors. Formed from the reaction between isophthalaldehyde and a helical amino acid-disubstituted ferrocene dihydrazide, these macrocycles contain from one to eight ferrocene moieties. The isolation of the four smallest of the macrocycles and their characterisation by UV-Vis, CD and NMR spectroscopy is described. The conformation of the macrocycles is explored, particularly with reference to the formation of a helical intramolecularly hydrogen-bonded structure. An investigation of the use of these macrocycles as anion-receptors shows that they are all effective hosts; the larger macrocycles show the highest affinities for anions. Studies using NMR spectroscopy suggest that the anion-recognition results primarily from the formation of multiple hydrogen bonds between the anions and the electropositive N–H protons of the acylhydrazones.
Introduction
We present here a series of new chirally-organised ferrocene-amino acid-based macrocycles synthesised by means of hydrazone formation from simple building blocks (Fig. 1(a) and (b)). Inspired by the known efficiency of neutral receptors that bind anions viahydrogen bonds,1 we chose to explore the anion-binding ability of our acylhydrazone-based macrocycles. While receptors based on amides,2pyrroles,3ureas,4ammonium5 and guanadinium6groups have received much attention, the use of hydrazone motifs to bind anions is relatively unexplored, although a small number of hydrazone-based photochemical sensors have recently been reported.7![(a) Dihydrazide building block Fc–[CO–Val–NHNH2]2 (V) and dialdehyde building block (I); (b) hydrazone-based macrocycles (VI)nnn can be synthesised from building blocks V and I under acidic conditions; (c) helicity and intramolecular hydrogen bonding in the Herrick conformation as viewed from above, and represented schematically.](/image/article/2011/SC/c1sc00168j/c1sc00168j-f1.gif) | ||
| Fig. 1 (a) Dihydrazide building block Fc–[CO–Val–NHNH2]2 (V) and dialdehyde building block (I); (b) hydrazone-based macrocycles (VI)nnn can be synthesised from building blocks V and I under acidic conditions; (c) helicity and intramolecular hydrogen bonding in the Herrick conformation as viewed from above, and represented schematically. | ||
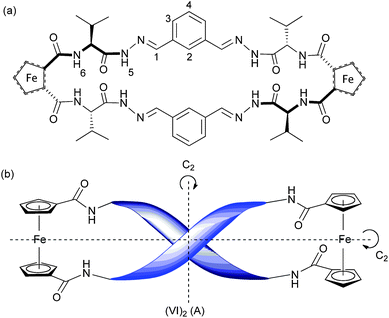
The use of ferrocene to induce β-sheet formation when incorporated into proteins, and the utility of ferrocene as a scaffold upon which to build β-sheet mimics, have been the subject of recent investigations.8 Inter-strand hydrogen bonds in amino acid-disubstituted ferrocenes can restrict the conformational freedom of the Cp rings to give well defined geometries. It is now well-established that amino acid disubstituted ferrocenes with the generic structure Fc–[CO–AA1–OR]2 or Fc–[CO–AA1–AA2–OR]2 (where AA1 is Val,9 Ala,10 Phe9a,10b,11 Gly,9c,11,12 His,13Cys(Me) or Cys(Bn)14) adopt a C2 symmetric helical conformation in chlorinated organic solvents (Fig. 1(c)). The chirality of the helix is determined by the chirality of the amino acid adjacent to the ferrocene moiety—L chirality leads to P helicity, D chirality leads to M helicity.10b In this “Herrick” conformation,9a the ferrocene substituents are positioned in an eclipsing 1,2′ conformation, the carbonyls adjacent to the Cp rings lying parallel to the rings and pointing in opposite directions; two hydrogen bonds may form between the NHs and carbonyls of AA1.
Ferrocene has previously featured in anion receptors, either as a structural component, exploited for its molecular hinge-like character, or to provide electrochemical sensing capability.15 We designed a ferrocene-based dihydrazide building block, Fc–[CO–Val–NHNH2]2 (V), that could react with isophthalaldehyde (I) to generate hydrazone macrocycles of various sizes (Fig. 1). We anticipated that the two valine moieties would play an important role in defining the conformational structure of the macrocycle as well as facilitating anion recognition by providing hydrogen-bond donors to complement the hydrazone moiety.
Our original aim was to explore the properties of the macrocycles generated using a dynamic combinatorial approach.16 To our surprise, however, the reaction between V and I to form hydrazones in the presence of acetic acid was not under thermodynamic control. Rather than generating a dynamic combinatorial library (DCL), a static mixture of macrocycles containing between 2 and 16 building blocks was formed. Elsewhere we will demonstrate how to transform such a static mixture of hydrazone macrocycles into a DCL. Using a dynamic combinatorial approach it is possible to rapidly expand the diversity of receptors to be studied and thereby identify unexpected but highly effective hosts.17 Here we investigate the helical structure of these ferrocene-amino acid-containing macrocycles and examine their capabilities as hydrazone-based anion receptors.
Synthesis and characterisation of macrocycles
Building block synthesis and characterisation
The ferrocene-dihydrazide building block V was synthesised in two steps (Scheme 1). 1,1′-Ferrocene dicarboxylic acid was coupled to two valine methyl esters, using standard peptide coupling conditions to form Fc–[CO–Val–OMe]2 (1) in 72% yield. Hydrazinolysis, initiated by stirring a solution of Fc–[CO–Val–OMe]2 (1) in hydrazine and MeOH, proceeded in 77% yield to generate the dihydrazide building block V as a pale yellow solid.![Synthesis of Fc–[CO–Val–NHNH2]2 (V): (a) H–Val–OMe, HOBt, EDC, Et3N, DMF, overnight, 72%; (b) N2H4·H2O, MeOH, 2 days, 77%.](/image/article/2011/SC/c1sc00168j/c1sc00168j-s1.gif) | ||
| Scheme 1 Synthesis of Fc–[CO–Val–NHNH2]2 (V): (a) H–Val–OMe, HOBt, EDC, Et3N, DMF, overnight, 72%; (b) N2H4·H2O, MeOH, 2 days, 77%. | ||
In Fig. 2 is shown the CD spectrum of Fc–[CO–Val–NHNH2]2 (V) and Fc–[CO–Val–OMe]21 in CHCl3/MeOH solution. It has previously been established that 1 has a Herrick-like P-helical conformation in solution9 and in the solid state,9b despite the fact that the NH–O interstrand contacts in the crystal structure are rather too long to be strictly described as hydrogen bonds. The P-helicity of the structure has been observed in crystal structures and is evidenced in solution by a distinctive positive CD signal at ∼485 nm.9c The similarity between the CD spectra of the methyl ester precursor 1 and the dihydrazide building block V suggest that the latter also has a P-helical structure in CHCl3/MeOH solution. The 1H-NMR spectrum of Fc–[CO–Val–NHNH2]2 (V) in CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3OD (96:4) (ESI† Fig. S2) shows four resonances for the ferrocene moiety and a single set of signals for the valine units, confirming that V has a C2 symmetry axis perpendicular to the Cp–Fe–Cp axis. The location of the amide N–H resonance at 7.98 ppm is relatively shielded for an amide proton, suggesting that these protons are engaged in hydrogen-bonding.8d The hydrazide NH protons appear not to be hydrogen bonded; the signal at 8.89 ppm is relatively shielded compared with hydrogen-bonded hydrazides in reported systems.18
:CD3OD (96:4) (ESI† Fig. S2) shows four resonances for the ferrocene moiety and a single set of signals for the valine units, confirming that V has a C2 symmetry axis perpendicular to the Cp–Fe–Cp axis. The location of the amide N–H resonance at 7.98 ppm is relatively shielded for an amide proton, suggesting that these protons are engaged in hydrogen-bonding.8d The hydrazide NH protons appear not to be hydrogen bonded; the signal at 8.89 ppm is relatively shielded compared with hydrogen-bonded hydrazides in reported systems.18
![CD spectra (2 mM, CHCl3 : MeOH (96 : 4)) of Fc–[CO–Val–OMe]2 (1) and Fc–[CO–Val–NHNH2]2 (V) (—), the positive absorbance at ∼485 nm indicates P-helicity at the ferrocene moiety.](/image/article/2011/SC/c1sc00168j/c1sc00168j-f2.gif) | ||
Fig. 2
CD spectra (2 mM, CHCl3:MeOH (96:4)) of Fc–[CO–Val–OMe]2 (1)  and Fc–[CO–Val–NHNH2]2 (V) (—), the positive absorbance at ∼485 nm indicates P-helicity at the ferrocene moiety. and Fc–[CO–Val–NHNH2]2 (V) (—), the positive absorbance at ∼485 nm indicates P-helicity at the ferrocene moiety. | ||
Macrocycle formation and isolation
Hydrazone-based macrocycles were generated by reacting Fc–[CO–Val–NHNH2]2 (V) (0.5 mM) with isophthalaldehyde (I) (0.5 mM) in CHCl3 in the presence of 0.5% acetic acid (v/v) (Scheme 2). The reaction mixture was analysed by HPLC after 4 days and showed the formation of several new species; these were identified using electrospray ionisation LC/MS to be the hydrazone macrocycles formed from the dihydrazide V and isophthalaldehyde (I) building blocks: (VI), (VI)22, (VI)33, (VI)44 ... (VI)88 (Fig. 3). | ||
| Scheme 2 Synthesis of macrocycles (VI), (VI)22, (VI)33 and (VI)44. | ||
![HPLC
chromatogram (290 nm) showing the distribution after four days of macrocycles formed from the reaction of Fc–[CO–Val–NHNH2]2 (V) with isophthalaldehyde (I).](/image/article/2011/SC/c1sc00168j/c1sc00168j-f3.gif) | ||
| Fig. 3 HPLC chromatogram (290 nm) showing the distribution after four days of macrocycles formed from the reaction of Fc–[CO–Val–NHNH2]2 (V) with isophthalaldehyde (I). | ||
Macrocycles
(VI), (VI)22, (VI)33 and (VI)44 were isolated using standard column chromatography on silica gel. The first column with a gradient eluent (CH2Cl2–CH2Cl2:MeOH (9:1)) enabled the separation of three fractions, containing (VI) and (VI)22 together, (VI)33 and (VI)44. A second column on the first fraction using an isocratic mixture of CH2Cl2:MeOH:Et3N (92.5:5:2.5) facilitated the separation of (VI) and (VI)22. Analysis of the isolated species by HPLC confirmed that the separations had been successful (ESI† Fig. S1). The use of standard column chromatography was an efficient means to isolate reasonable quantities of macrocycles compared with preparative-scale HPLC, which is more commonly employed for the separation of such large and similar macrocycles.
Characterisation by UV/Vis and CD spectroscopy
The UV/Vis spectra of solutions of macrocycles (VI), (VI)22, (VI)33 and (VI)44 in the region 240 nm to 400 nm are shown in Fig. 4(a). The shape of the absorbance bands for macrocycles (VI)22, (VI)33 and (VI)44 are essentially identical, although the extinction coefficients per hydrazone linkage vary. The shape of the absorbance band for (VI) is, however, distinctly different; the presence in the spectrum of vibrational detail might imply that the structure is very rigid.19 The hydrazone linkages in macrocycle (VI) may exhibit differing electronic properties due to the presence of ring strain or different cis/trans and/or E/Z isomers.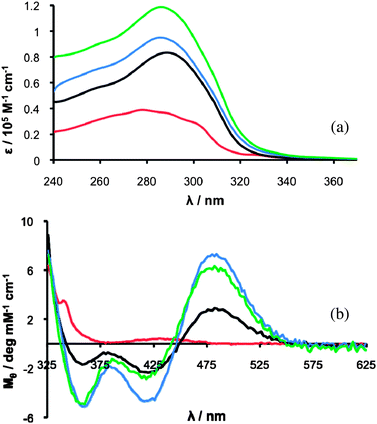 | ||
| Fig. 4 (a) UV/Vis spectra (CHCl3:MeOH (96:4)) of (VI) (25 μM, red trace), (VI)22 (13 μM, black trace), (VI)33 (7.9 μM blue trace) and (VI)44 (6.3 μM green trace). (b) CD spectra in CHCl3:MeOH (9:1) of (VI) (1.0 mM, red trace), (VI)22 (0.50 mM, black trace), (VI)33 (0.33 mM, blue trace) and (VI)44 (0.25 mM, green trace). | ||
The CD spectra of macrocycles (VI)22, (VI)33 and (VI)44 show the distinctive positive CD signal, at ∼485 nm, observed for Fc–[CO–Val–OMe]2 (1) and Fc–[CO–Val–NHNH2]2 (V) (Fig. 4(b)). The absence of a positive absorbance at ∼485 nm in the CD spectrum of (VI) indicates that, unlike the larger macrocycles, it does not show the sort of P-helicity at the ferrocene moiety that is induced by a Herrick hydrogen bonding pattern. Most likely, the macrocycle is too small and not flexible enough to be able to adopt a Herrick conformation. Macrocycle (VI) is distinguished from the larger macrocycles by its differing UV/Vis and CD spectra, a distinction that is also reflected in its lower stability, as described below.
Characterisation by NMR spectroscopy
The 1H-NMR spectrum of (VI) recorded at room temperature showed sharp signals in the aromatic region, some broad signals in the ferrocene region (4–5 ppm) but a complete absence of α and β amino acid proton signals (ESI† Fig. S5). When the temperature was lowered, however, it was possible to observe two α and two β protons, two γ signals, two amide N–H proton (H6/H6′) signals as well as two hydrazone NH protons (H5/H5′) (Fig. 5). A COSY experiment at low temperature enabled the assignment of two sets of peaks for two non-equivalent valine units (distinguished by markings in red and blue in Scheme 3 and Fig. 5) present in equal concentrations at varying temperatures. The exchange between the two isomers was confirmed by a NOESY experiment, which clearly showed exchange cross-peaks between the two α-proton signals and the two β-proton signals (ESI† Fig. S6(a)). | ||
| Scheme 3 Equilibrium between isomers of macrocycle (VI). | ||

We therefore deduced that there must be one unsymmetrical structure, the red and blue peaks corresponding to the different sides of the macrocycle, which flips between two identical conformations, a phenomenon that is comparable with the conformational flipping of a syn-1,4-disubstituted cyclohexane. The valine units are chemically inequivalent but they exchange such that each valine moiety spends half of its time in each of the two chemical environments (Scheme 3).
In the NMR spectrum at 278 K (Fig. 5), the amide NH proton resonances (H6 and H6′) are located at 7.33 ppm (red) and 10.00 ppm (blue); this difference, and the particularly downfield shift to 10.00 ppm, suggest that at any given moment one of the amide NH protons (H6/H6′) is engaged in strong hydrogen bonding, while the other is not. Meanwhile the imine proton signals (H1 and H1′) are completely overlapped, and the hydrazone NH protons (H5 and H5′) and the aromatic protons (H3 and H3′) both have similar chemical shifts. This suggests that the chemical environment on either side of the isophthalaldehyde moiety are almost identical. We conclude that the imine must be parallel to the ArC–H2 bond, rather than parallel to the ArC–H3 bond because the H2 resonance is positioned unusually further downfield than the imine proton signal (H1/H1′); this may be explained by the fact that in this position H2 would lie in the deshielding region of the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds and potentially hydrogen bond to the two imine nitrogens. The observation of nOe cross peaks between the imine H1/H1′ signal and both hydrazone NH protons (H5 and H5′), and not between H5 and H5′ and H2 (ESI† Fig. S6(b)) suggested that both imines are in an E conformation with both H5 and H5′ pointing to the outside of the macrocycle as shown in Scheme 3. On the basis of the NMR spectra, therefore, we propose that the macrocycle (VI) exists in a “van Staveren” conformation, previously only observed in the solid state structure of Fc–[CO–Phe–OMe]2,11 in which the carbonyls adjacent to the Cp rings point in the same direction and a single interstrand hydrogen bond forms between the amide NH adjacent to one ring, and the carbonyl on the other.
N double bonds and potentially hydrogen bond to the two imine nitrogens. The observation of nOe cross peaks between the imine H1/H1′ signal and both hydrazone NH protons (H5 and H5′), and not between H5 and H5′ and H2 (ESI† Fig. S6(b)) suggested that both imines are in an E conformation with both H5 and H5′ pointing to the outside of the macrocycle as shown in Scheme 3. On the basis of the NMR spectra, therefore, we propose that the macrocycle (VI) exists in a “van Staveren” conformation, previously only observed in the solid state structure of Fc–[CO–Phe–OMe]2,11 in which the carbonyls adjacent to the Cp rings point in the same direction and a single interstrand hydrogen bond forms between the amide NH adjacent to one ring, and the carbonyl on the other.
The NMR experiments performed on macrocycle (VI)22 revealed that in a CDCl3:CD3OD (9:1) solution (VI)22 exists as three different isomers: one with D2 symmetry (from here on referred to as A and coloured in blue), one with C2 symmetry (B, red) and one with C1 symmetry (C, green). Fig. 6 shows the aromatic region of the spectrum in a variable temperature NMR experiment. It can be seen that the concentrations of the different isomers vary with temperature. Isomer A (blue), present in higher concentrations at low temperature is the most enthalpically stable isomer; isomer B (red) is more entropically stable. The fact that exchange between the isomers takes place readily at room temperature suggests that they are not geometric E/Zimine isomers, but rather conformational isomers due to cis/transamides or another source of restricted rotation.
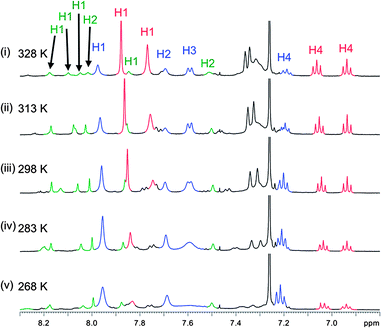 | ||
| Fig. 6 Partial 1H-NMR spectra of (VI)22 in CDCl3:CD3OD (9:1) at (i) 268 K, (ii) 283 K, (iii) 298 K, (iv) 313 K and (v) 328 K. | ||
COSY and NOESY experiments were used to assign the imine protons (H1) and aromatic protons (H2, H3 and H4) (ESI† S2.3). For isomer A, there is only one imine proton (H1) signal, although there are four imine protons in macrocycle (VI)22. Given the presence of chiral amino acids in the macrocycle, this is only possible if there are two C2 symmetry axes running through the molecule, one parallel to the Cp–Fe–Cp axis, through H4 and H2 and one perpendicular to the Cp–Fe–Cp axis, running through both Fe atoms. For isomer B, there are two imine proton (H1) signals, which is only possible if there is one C2 symmetry axis running through the molecule. Since two signals for H4 can clearly be discerned, it was concluded that the C2 symmetry axis runs parallel to the Cp–Fc–Cp axis through H4 and H2. For isomer C, four different signals are seen for the four different imine protons (H1) and there are no symmetry axes in this isomer.
In the NOESY spectrum of (VI)22 (ESI† Fig S9), there are nOe crosspeaks between the signals of H1 and H2 of the D2 symmetric isomer A (blue), suggesting that in this isomer the imine CN bonds are parallel to the ArC–H3 bond, the opposite of what was observed for the smaller macrocycle (VI). Additionally, the H2 signal is no longer significantly shifted downfield of the H1 signal, as was seen for (VI). Meanwhile, nOe cross peaks were observed between imine H1 and hydrazone NH(5) indicating the close proximity of these two protons and therefore that the imines in isomer A are in an E configuration. The structure of isomer A of (VI)22 shown in Fig. 7 is proposed on the basis of these NMR constraints, the D2 symmetry of the molecule, and the evidence of P-helicity and a Herrick conformation seen in the CD spectrum of (VI)22. Unfortunately it was not possible to investigate the structure of (VI)33 and (VI)44 using NMR spectroscopy due to the complexity of their spectra resulting from the presence of multiple isomers (ESI†, Fig. S10 and Fig. S11).
 | ||
| Fig. 7 Proposed structure of isomer A of (VI)22: (a) shown from above and (b) represented schematically from a side view. | ||
Macrocycle stability
Hydrazones, despite their electropositive imine and N–H protons, have in only a few cases been investigated as potential anion receptors,7 which may be in part due to their reported instability and acid lability. While stable under neutral and basic conditions, hydrazones often readily undergo exchange in the presence of acid and thus have been exploited in the field of dynamic combinatorial chemistry.17,20We found, however, that a solution of (VI)22 (0.5 mM) in CHCl3 containing 0.5% acetic acid, underwent no exchange and was stable for at least 12 days. While the individual hydrazone linkages in (VI)22 may be labile, the high thermodynamic stability of the macrocycle—possibly due to intramolecular hydrogen bonding—compared with the products of hydrolysis or an exchange reaction, means that (VI)22 is kinetically trapped and therefore stable even in the presence of acid. In other words, even if the ring opens by fission at one hydrazone linkage, the resulting linear tetramer will snap closed before any intermolecular reaction can take place. As a consequence, the use of (VI)22 as an anion receptor is not restricted to basic and neutral environments.
Macrocycle (VI), in contrast, was unstable in the presence of even small quantities of acid, readily undergoing exchange to generate a mixture of higher order macrocycles.‡ The reaction of Fc–[CO–Val–NHNH2]2 (V) and isophthalaldehyde (I) in the presence of 0.5% acetic acid in CHCl3 to generate macrocycles (VI), (VI)22, (VI)33 and (VI)44 was monitored over a period of 70 h by HPLC (Fig. 8). The reaction profile shows that (VI) is the fastest forming macrocycle, but after approximately 4 h it reaches its maximum concentration. The larger macrocycles, initially slow to form, continue to increase in concentration for up to at least 20 h. The rate of formation of the larger macrocycles appears to be dependent on the concentration of (VI), which suggested that (VI) is an intermediate in their synthesis. The relative instability of (VI) is presumably a consequence of the smaller ring size and differing hydrazone conformation.
![Reaction profile for the synthesis of macrocycles from Fc–[CO–Val–NHNH2]2 (V) and isophthalaldehyde (I) monitored by HPLC: (VI) (-□-); (VI)22 (-△-); (VI)33 (-▲-); (VI)44 (-■-).](/image/article/2011/SC/c1sc00168j/c1sc00168j-f8.gif) | ||
| Fig. 8 Reaction profile for the synthesis of macrocycles from Fc–[CO–Val–NHNH2]2 (V) and isophthalaldehyde (I) monitored by HPLC: (VI) (-□-); (VI)22 (-△-); (VI)33 (-▲-); (VI)44 (-■-). | ||
Anion binding studies
UV-Vis spectroscopy
The anion-binding ability of macrocycles (VI), (VI)22, (VI)33 and (VI)44 was studied using UV-Vis spectroscopy and NMR spectroscopy. UV-Vis spectroscopy titrations were used to determine the stoichiometry and association constants (K) for the interactions between macrocycles (VI), (VI)22, (VI)33 and (VI)44 and Bu4NHSO4, Bu4NH2PO4, Bu4NNO3, Bu4NOAc, BnEt3NBr, BnEt3NBF4 and BnEt3NCl in CHCl3:MeOH (96:4) solution, and in some cases also in CHCl3 solution (Table 1).
:1) determined from UV/Vis titrations in CHCl3:MeOH (96:4) and CHCl3 solutions (errors < 12%)
| Guest | 1:1 Binding Constant K/103 M−1 |
|||||
|---|---|---|---|---|---|---|
| (VI) | (VI)22 | (VI)33 | (VI)44 | |||
| CHCl3 + MeOH | CHCl3 | CHCl3 + MeOH | CHCl3 | CHCl3 + MeOH | CHCl3 + MeOH | |
| Bu4NH2PO4 | 11 | 26 | 12 | — | 26 | 43 |
| Bu4NHSO4 | 5 | 11 | 15 | 32 | 14 | 21 |
| BnEt3NBF4 | 4 | 6 | 12 | 27 | 28 | 41 |
| Bu4NOAc | 5 | — | 12 | — | 21 | 30 |
| Bu4NNO3 | 5 | 4 | 10 | 36 | 26 | 48 |
| BnEt3NCl | 3 | 4 | 14 | 37 | 20 | 47 |
| BnEt3NBr | 7 | 4 | 8 | 22 | 17 | 37 |
The binding isotherms fitted well to a 1:1 binding model. Furthermore, Job plots prepared for selected macrocycles and anions clearly confirmed that each macrocycle bound one anion. Fig. 9 displays the binding isotherm and Job plot obtained for the interaction between (VI)22 and Bu4NHSO4 as an example.
 | ||
| Fig. 9
UV/Vis
titration binding isotherm and Job plot (inset) for the interaction between Bu4NHSO4 and (VI)22 in CHCl3:MeOH (96:4). | ||
All of the macrocycles displayed a reasonably strong affinity for each of the anions. In all cases, except Bu4NHSO4, the association constants increased with the increasing size of the macrocycle. It may be that the greater flexibility of (VI)33 and (VI)44 allows them to adapt their shape to optimise favourable intermolecular interactions with the anion or that the presence of multiple binding sites on these large macrocycles increases the probability of a favourable interaction. There was little selectivity seen for particular anions by the macrocycles, with the exception of (VI), which bound Bu4NH2PO4 with a significantly higher binding constant compared with the other anions. By contrast, (VI)22 bound most strongly to Bu4NHSO4, which also showed a significantly lower affinity than the other anions for the larger macrocycles. The interactions between (VI) and Bu4NH2PO4, and (VI)22 and Bu4NHSO4 were further studied, using NMR spectroscopy.
Study of anion binding by (VI) using NMR spectroscopy
A solution of (VI) in CDCl3:CD3OD (96:4) (4 mM) was titrated with Bu4NH2PO4 and the changes in the 1H-NMR spectrum were monitored (ESI† Fig. S12). A very small downfield shift and broadening of the imine H1/H1′ proton resonance was observed, while no other significant shifts were seen. The NH resonances, could not be observed at room temperature as they are in fast exchange with protons from the CD3OD. However, it was possible to judge their involvement in the binding of H2PO4− by comparing their chemical shifts at 268 K in the absence, and the presence of six equivalents of Bu4NH2PO4. It was found that the hydrazone NH protons (H5 and H5′) experienced downfield shifts of 0.24 ppm and 0.72 ppm, while the amide NH protons (H6 and H6′) did not experience any significant shift, suggesting that the hydrazone NH protons (H5/H5′) hydrogen bond to the anionic guest while the amide NH protons (H6/H6′) do not.
The proposed structure of (VI) (Scheme 3) shows the imine protons (H1 and H1′) and the hydrazone NH protons (H5 and H5′) within close proximity and orientated to the exterior of the macrocycle, with the imine nitrogen pointing inwards. The very small downfield shift seen for imine protons (H1/H1′) is inconsistent with the formation of hydrogen bonds to the anion. More likely, these protons are not directly involved in binding, but rather are deshielded due to a shift of electron density towards the nitrogens, which engage in hydrogen bonding with the protons of the H2PO4−. A small rotation around the N–N bonds could allow the hydrazone NH protons (H5 and H5′) to point sufficiently towards the cavity to hydrogen bond to oxygens from H2PO4− positioned in the cavity the macrocycle. The amide NH protons (H6 and H6′), meanwhile are not involved in binding the anion.
Study of anion binding by (VI)22 using NMR spectroscopy
When a solution of (VI)22 in CDCl3:CD3OD (9:1) (4 mM) was titrated with Bu4NHSO4, small shifts in peaks belonging to isomers A (D2), B (C2) and C (C1) were observed, indicating that each isomer binds the anion to some extent (Fig. 10). The largest shifts were seen for isomers A (blue) and C (green). Different H1 protons of isomer C shift up and down field, suggesting that some significant rearrangement of the macrocycle is required in order to effectively bind HSO4−. In the course of the titration, the proportion of isomer A in the mixture increases slightly at the expense of isomer B, indicating that isomer A binds the anion more strongly than isomer B.
 | ||
| Fig. 10 Partial 1H-NMR spectra of (VI)22 (2.0 mM) in CDCl3:CD3OD (9:1) at 298 K in the presence of Bu4NHSO4: (i) 0 mM, (ii) 8 mM, (iii) 18 mM, (iv) 25 mM, (v) 34 mM, (vi) 42 mM and (vii) 50 mM. | ||
The most significant changes observed in the spectrum of isomer A during the titration were the downfield shift of imine protons (H1), the upfield shift of H2 and the downfield shift of hydrazone NH protons (H5). It was not possible to observe the amide proton signals in this solution containing 10% CD3OD. It can therefore only be assumed that the macrocycle binds H2SO4−via hydrogen bonding with the imine H1 protons, and possibly also hydrogen bonding with the hydrazone NHs in a manner that maintains the D2 symmetry of the macrocycle.
Conclusions
The reaction of two building blocks—a dihydrazide, Fc–[CO–Val–NHNH2]2 (V) and a dialdehyde, isophthalaldehyde (I)—under acidic conditions has given rise to a series of new ferrocenophanes incorporating from one up to eight ferrocene moieties. The acylhydrazones that link the alternating building blocks may be exploited for anion recognition. Macrocycles (VI)22(VI)33 and (VI)44 exhibit P-helical structures in solution and show CD spectra consistent with a Herrick conformation. Macrocycle (VI)22 exists in solution as a mixture of three different conformers of varying symmetry—D2, C2 and C1—in slow exchange with one another and present in an approximately 1:2:1 ratio. The most enthalpically stable conformer is the most symmetrical conformer (D2). Macrocycle (VI) exists in solution as an unsymmetrical macrocycle that exchanges slowly on an NMR timescale between two identical conformations. It is small and unstable and sterically prevented from adopting a Herrick structure, generating instead a van Stavaren conformation. The macrocycles each form 1:1 complexes with anions in CHCl3:MeOH solution. Despite the generally-assumed instability of acylhydrazones, macrocycle (VI)22 proved to be resistant to hydrolysis or exchange even under acidic conditions, illustrating how acylhydrazones can form stable molecules and are therefore deserving of further exploration as anion receptors.
We thank the EPSRC and the Gates Cambridge Trust for financial support, and Dr Ana Belenguer for maintaining the HPLC facility.
Notes and references
- (a) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, U.K., 2006 Search PubMed; (b) S. Kubik, C. Reyheller and S. Stuwe, J. Inclusion Phenom. Macrocyclic Chem., 2005, 52, 137–187 CrossRef CAS; (c) K. H. Choi and A. D. Hamilton, Coord. Chem. Rev., 2003, 240, 101–110 CAS.
- (a) S. Kubik, Chem. Soc. Rev., 2009, 38, 585–605 RSC; (b) S. O. Kang, M. A. Hossain and K. Bowman-James, Coord. Chem. Rev., 2006, 250, 3038–3052 CrossRef CAS; (c) C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77–99 CAS.
- (a) J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17–55 CAS.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- (a) E. Garcia-Espana, P. Diaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952–2986 CrossRef CAS.
- (a) C. Schmuck, Coord. Chem. Rev., 2006, 250, 3053–3067 CrossRef CAS; (b) M. D. Best, S. L. Tobey and E. V. Anslyn, Coord. Chem. Rev., 2003, 240, 3–15 CAS.
- (a) Y.-M. Zhang, Q. Lin, T.-B. Wei, X.-P. Qin and Y. Li, Chem. Commun., 2009, 6074–6076 RSC; (b) J. Shao, Y. Qiao, H. Lin and H. Lin, Spectrochim. Acta, Part A, 2009, 71, 1736–1740 Search PubMed; (c) Y.-H. Qiao, H. Lin and H.-K. Lin, J. Inclusion Phenom. Macrocyclic Chem., 2007, 59, 211–215 CrossRef CAS.
- (a) A. Lataifeh, S. Beheshti and H.-B. Kraatz, Eur. J. Inorg. Chem., 2009, 2009, 3205–3218 Search PubMed; (b) N. Metzler-Nolte and M. Salmain, in Ferrocenes: Ligands, Materials and Biomolecules, (ed. P. Štěpnička), John Wiley and Sons, Chichester, U.K., 2008 Search PubMed; (c) T. Moriuchi and T. Hirao, Acc. Chem. Res., 2010, 43, 1040–1051 CrossRef CAS; (d) S. I. Kirin, H.-B. Kraatz and N. Metzler-Nolte, Chem. Soc. Rev., 2006, 35, 348–354 RSC.
- (a) R. S. Herrick, R. M. Jarret, T. P. Curran, D. R. Dragoli, M. B. Flaherty, S. E. Lindyberg, R. A. Slate and L. C. Thornton, Tetrahedron Lett., 1996, 37, 5289–5292 CrossRef CAS; (b) M. Oberhoff, L. Duda, J. Karl, R. Mohr, G. Erker, R. Frohlich and M. Grehl, Organometallics, 1996, 15, 4005–4011 CrossRef CAS; (c) S. I. Kirin, U. Schatzschneider, S. D. Koster, D. Siebler and N. Metzler-Nolte, Inorg. Chim. Acta, 2009, 362, 894–906 Search PubMed.
- (a) A. Nomoto, T. Moriuchi, S. Yamazaki, A. Ogawa and T. Hirao, Chem. Commun., 1998, 1963–1964 RSC; (b) S. I. Kirin, D. Wissenbach and N. Metzler-Nolte, New J. Chem., 2005, 29, 1168–1173 RSC.
- D. van Staveren, T. Weyhermuller and N. Metzler-Nolte, Dalton Trans., 2003, 210–220 RSC.
- F. E. Appoh, T. C. Sutherland and H.-B. Kraatz, J. Organomet. Chem., 2004, 689, 4669–4677 CrossRef CAS.
- S. Chowdhury, G. Schatte and H.-B. Kraatz, Eur. J. Inorg. Chem., 2006, 988–993 CrossRef CAS.
- X. de Hatten, T. Weyhermuller and N. Metzler-Nolte, J. Organomet. Chem., 2004, 689, 4856–4867 CrossRef CAS.
- (a) S. R. Bayly, P. D. Beer and G. Z. Chen, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley and Sons, Chichester, U.K., 2008 Search PubMed; (b) P. D. Beer and S. R. Bayly, Top. Curr. Chem., 2005, 255, 125–162 CAS; (c) P. D. Beer, Z. Chen, A. J. Goulden, A. Graydon, S. E. Stokes and T. Wear, Chem. Commun., 1993, 1834–1836 RSC; (d) P. D. Beer, A. R. Graydon, A. O. M. Johnson and D. K. Smith, Inorg. Chem., 1997, 36, 2112–2118 CrossRef CAS; (e) F. Oton, A. Tarraga and P. Molina, Org. Lett., 2006, 8, 2107–2110 CrossRef CAS.
- (a) J. N. H. Reek and S. Otto, ed., Dynamic Combinatorial Chemistry, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed; (b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- S. R. Beeren and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3804–3807 Search PubMed.
- X. Zhao, X.-Z. Wang, X.-K. Jiang, Y.-Q. Chen, Z.-T. Li and G.-J. Chen, J. Am. Chem. Soc., 2003, 125, 15128–15139 CrossRef CAS.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, California, 2006 Search PubMed.
- (a) R. T. S. Lam, A. M. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS; (b) V. T. Bhat, A. M. Caniard, T. Luksch, R. Brenk, D. J. Campopiano and M. F. Greaney, Nat. Chem., 2010, 2, 490–497 CrossRef CAS; (c) J. M. Klein, V. Saggiomo, L. Reck, M. McPartlin, G. D. Pantoş, U. Lüning and J. K. M. Sanders, Chem. Commun., 2011, 47, 3371–3373 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional NMR spectra, UV/Vis, binding isotherms and Job plots. See DOI: 10.1039/c1sc00168j |
| ‡ Samples of (VI) reacted to form a mixture of larger macrocycles when exposed to the trace amounts of acid present in commercial CDCl3. Additionally, a dilute sample of (VI) (2.5 μM) in CHCl3:MeOH over a period of weeks reacted to form a mixture of macrocycles. |
| This journal is © The Royal Society of Chemistry 2011 |