Self-assembly of a family of macrocyclic polyoxotungstates with emergent material properties†
Jing
Gao
,
Jun
Yan
,
Scott G.
Mitchell
,
Haralampos N.
Miras
,
Antoine G.
Boulay
,
De-Liang
Long
* and
Leroy
Cronin
*
WestCHEM, School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow G12 8QQ, UK. E-mail: deliang.long@glasgow.ac.uk; lee.cronin@glasgow.ac.uk; Web: http://www.croninlab.com Fax: +44 (0)141 330 4888; Tel: +44 (0)141 330 6650
First published on 31st May 2011
Abstract
Using a time-dependent synthetic approach, the construction of a new class of macrocyclic tungstotellurite heteropolyacid cluster-squares, [W28Te8O112]24− (1a), [W28Te9O115]26− (2a) and [W28Te10O118]28− (3a) has been achieved. Isolation of these architectures is facilitated by the tellurite anions, which act as templates within structural building units and linkers between them, as well as pendant ligands which subsequently control inter-cluster aggregation, thereby defining a new architectural principle in the assembly of polyoxotungstate clusters. ESI-MS studies on these assemblies provide insights into the self-assembly process, distinguishing the different structural roles between the templating, linking and pendant tellurium-based anions. Further, cation exchange reactions on the crystalline forms of the macrocyclic POMs cause a morphological transformation resulting in the emergence of microtubular architectures with vast aspect ratios thereby opening the way to fabricate new material architectures capable of exploiting the properties of HPAs in applications such as ion exchange, catalysis and sensing.
Introduction
Polyoxometalates (POMs) are discrete polyanionic metal oxide clusters based on building blocks of general formula {MOx}n, where M = Mo, W, V, and Nbetc. and x = 4–7.1 Not only do POMs possess a remarkable range of structures, the molecular and electronic versatility affords them an almost unmatched range of physical and chemical properties, thereby permitting their applications in many fields: from catalysis and photochemistry, to medicine and materials science.2 Heteropolyanions (HPAs), which contain heteroatoms such as Si, P or Geetc., represent by far the most versatile subset of POM clusters. Although POM chemistry has undergone a rapid development during the previous century, the assembly process of HPAs is still not well understood since most of the HPAs are isolated in “one-pot” reactions. Furthermore, to date, the number of large macrocyclic HPAs which contain sizeable, largely unoccupied, cavities is limited to only three examples.3–6 Perhaps the most well-known example of an HPA macrocycle is the {P8W48} polyanion which displays versatile host–guest chemistry.3,4 Despite recent research however, the isolation of nonconventional anionic template-containing macrocyclic HPAs is surprisingly rare and only two clusters, i.e., {As4W40}5 and {Sb8W36},6 have been isolated in 1978 and 1997, respectively. In fact, the embedding of the lone electron pair containing anionic templates can restrict the formation of Keggin-type clusters and then significantly vary the self-assembly process in solution, allowing the discovery of intrinsically new building blocks as well as the isolation of novel clusters such as the trimetric [W21O77Sb6(SbO3)3Na]18−.7 By adopting this route we have been able to explore and exploit the hetero-template effect in HPAs e.g. in the development of novel Dawson type clusters embedded with electronically active anions,8–10 and even gigantic saddle shaped clusters.11 Therefore we realised that there is a real opportunity to design and exploit new POM architectures that combine building blocks, both templated and linked by heteroanions, to develop macrocyclic systems as these should have novel architectures and potential supramolecular properties (e.g. guest binding), as well as the useful applicable properties of HPAs especially if they could be fabricated into novel material architectures. These prospects have been limited since the fabrication of such inorganic macrocycles into robust molecular-based materials is complicated by the difficulty in processing complex inorganic materials, and this is in stark contrast to organic functional materials.Herein we report an unprecedented family of clusters that fall into the new sub-class of macrocyclic HPA whereby the structures are directed by the tellurium based heteranion templates and linkers, see Fig. 1.
 | ||
| Fig. 1 Representation of the inner ‘square’ framework of the cluster anions 1a, 2a and 3a. A side view is shown on the left and a top view on the right. The templating and linking tellurium ions are highlighted by the pink and orange arrows respectively. All cations and solvent water molecules are omitted for clarity. Polyhedra: WO6; purple spheres: oxygen; blue spheres: Te; orange spheres: W. Dashed black lines represent weak interactions between Te⋯O. | ||
These clusters are: [W28Te8O112]24− (1a), [W28Te9O115]26− (2a) and [W28Te10O118]28− (3a), which crystallize as (C2H8N)6Na18[W28Te8O112]·53H2O (1), Na26[W28Te9O115]·37H2O (2) and Na28[W28Te10O118]·45H2O (3), respectively.12 Not only does this family expand the rare class of macrocyclic HPAs, we show that these macrocycles can ‘ligate’ additional {TeO3} units; one for compound 2 and two such units for compound 3. As a result, within compounds 2 and 3, the isolated [Te2O6]4− dimer moiety {O2Te-O-TeO3} has been observed within a cluster for the first time. Furthermore, we show the assembly of these macrocylic HPA clusters is uniquely controlled by a time-dependent self-assembly process that requires the continuous addition of tellurite anions to the parent polyanion structure during the formation of 1, 2 and 3. The cluster structures and assembly process are also studied by high resolution ESI-MS studies which provide important insights into the building blocks and thus the possible mechanism of self-assembly of the clusters. Finally, we show that in the solid state, compounds 1–3 can be transformed from crystalline samples into novel materials comprising hollow microtubular architectures with vast aspect ratios. These are fabricated by simple cation exchange reactions and open up ways to develop novel solid state devices that can exploit the intrinsic properties of HPAs, namely ion exchange, catalysis, and sensing.
Results and discussion
Structural analysis
Compound (C2H8N)6Na18[W28Te8O112]·53H2O 1 (Fig. 2) was synthesized by refluxing an aqueous solution of sodium tungstate, sodium tellurite and dimethylamine hydrochloride at pH 7.0 for half an hour whilst stirring. Subsequent crystallization over a three-week period yielded a hydrated sodium salt in the orthorhombic space groupPmmn. Single crystal X-ray structural analysis showed the overall unit to be a “saddle shaped” polyanionic cluster [W7O25(TeO3)Te]424− (1a) constructed from four TeIV linkers and four equivalent {W7O25(TeO3)} fragments. The TeIV heteroatom templates within the fragments have an average Te–O bond length of 1.888(1) Å, a little shorter than the TeIV linkers which are 1.997(3) Å in length.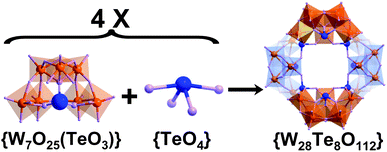 | ||
| Fig. 2 Scheme of the formation of compound 1 where four {W7O25(TeO3)} fragments combine with four {TeO4} linkers forming the square saddle-shaped compound {W28Te8O112}. All cations and solvent water molecules are omitted for clarity. Polyhedra: WO6; purple spheres: oxygen; blue spheres: Te; orange spheres: W. | ||
Each {W7O25(TeO3)} building block fragment can be considered as an assembly of three types of lower nuclearity building blocks: one TeIV, one tritungstate {W3} fragment made of three edge-shared [WO6] octahedra and two ditungstate {W2} subunits made of two edge-shared [WO6] octahedra. The {W3} unit and the TeIV are connected via a μ4-oxo bridge, which altogether then connects to the two ditungstate {W2} subunits through four μ2-oxo bridges and two μ3-oxo ligands, forming the principle {W7O25(TeO3)} fragment. The saddled-shaped cluster 1a is formed by linking the four building blocks {W7O25(TeO3)} by four TeIV ions. It is noteworthy that the tellurium centres in polyanion 1a exert two functions at the same time: four TeIV ions are located between pairs of the four {W7O25(TeO3)} fragments as linkers while four others are embedded within the {W7O25(TeO3)} fragments themselves, effectively acting as templates. This is interesting since this work establishes the idea that the use of hetero-anions as both templates and linkers can be exploited as a major new architectural principle in the design of nanoscale polyoxotungstates since normally they are only involved as central templating units.6,13
The structures of compound 2 and 3 are very similar to that of compound 1, differing only in the number of additional TeO32− units coordinated to the inside circles of the “saddle” (Figs. 3 and 4). The average Te–O bond lengths of the hanging TeO32− unit in compounds 2 and 3 are 1.920(9) Å and 1.958(5) Å respectively, longer than that of the tellurium ions encapsulated in {W7O25(TeO3)} fragments (which are 1.880(7) Å for 2 and 1.902(6) Å for 3). The average Te–O bond lengths of the TeIV linkers in compound 2 and 3 are 2.00(5) Å and 2.010(4) Å, respectively, a little longer than that of compound 1.
 | ||
| Fig. 3 Representation of the structure of compound 2 {W28Te9O115} (left) and compound 3 {W28Te10O118} (right). All cations and solvent water molecules are omitted for clarity. Polyhedra: WO6; purple spheres: oxygen; blue spheres: Te; orange spheres: W; black dashed lines: weak interactions between Te and oxygen atoms from cluster; green dashed line: weak interactions between Te templates in compound 3 and oxygen atoms from solvent water. | ||
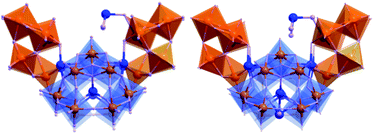 | ||
| Fig. 4 From left to right: side views of compound 2 and 3. | ||
These pendant TeO32− units mean that compounds 2 and 3 have some additional features to those of compound 1. One important feature is the presence of one [Te2O6]4− dimer in compound 2 and two such dimers in compound 3. These [Te2O6]4− dimers are the result of oxygen sharing between the template TeO32− embedded within {W7O25(TeO3)} fragments and a pendant TeO32−. Such units have only been reported twice before14,15 as part of chain polymer structures; but never before as isolated units as we show here. An explanation for the isolated Te–O–Te bonds we observe in compounds 2 and 3 could be unique to the POM, which is both highly charged and exerts a significant stereochemical effect, both of which may prevent additional TeO32− units from coordinating and thus preventing Te–O–Te polymerisation.
Synthesis
The syntheses of 1–3 were performed at neutral pH, showing that highly acidic conditions are not always essential for the isolation of high nuclearity polyoxometalate architectures. It is also noteworthy that the syntheses of compounds 1–3 are almost identical, differing only in the length of reflux time (from 0.5 h to more than 28 h). If the pH neutral solutions of the reactants were left to reflux for less than 2 h, the principle product isolated was compound 1; however, it was found that paratungstate B {H2W12O42} co-crystallized as a by-product following further evaporation of the mother liquor. Following many experiments, we have been unable to isolate single crystals of compound 1 without the presence of potassium or dimethylammonium (DMA) cations. Without the presence of these cations in solution, the only observed crystalline product was always {H2W12O42}. Further, if the reflux time was more than 2 h but less than 24 h, compound 2 would first crystalize out as pure product; however, if lengthy evaporation was permitted, compound 3 would then appear as the minor product. Thus, to obtain 2 with high purity these crystals were harvested early at the expense of the yield of product. Importantly, for reflux times longer than 24 h, compound 3 was isolated as the main product and as the only product for reflux times of more than 28 h. These aforementioned reflux time-scales demonstrate that the formation of compounds 1–3 is controlled both by the time of reaction and therefore the availability of tellurite anions in solution.Interactions between the tellurite-based hetero-anions
One rather novel aspect of the structures is the large number of intra- and inter-molecular interactions within/between the cluster structures, which are the reason why TeIV has such abundant coordination modes in these structures (Fig. 5). The intra-interactions result from weak O⋯Te coordinative interactions between the Te-based hetero-anion linkers and oxygen atoms from {W7O25(TeO3)} fragments (shown as black dashed lines in Fig. 3), which exists in all the three compounds. Further, compound 3 is unique in that the TeIV atoms contained within the {W7O25(TeO3)} fragments are involved in weak interactions with oxygen atoms from solvent water molecules present in the cluster cavity (shown as green dashed lines in Fig. 3). | ||
| Fig. 5 Overview of the TeIV coordination modes present in compounds 1–3. Numerical values correspond to distances (Å) between Te and O in weak, non-bonding Te⋯O interactions. Purple spheres: oxygen; blue spheres: Te linker; orange spheres: Te template; green spheres: Te pendant; black dashed lines: weak interactions between Te and oxygen atoms from cluster; green dashed line: weak interactions between Te templates in compound 3 and oxygen atoms from solvent water. | ||
Also, some weak inter-molecular interactions result from the pendent TeIV-based units and oxygen atoms from neighbouring {W7O25(TeO3)} fragments (black dashed lines in Fig. 6). Due to the presence of the pendant TeO32−, these inter- molecular interactions only exist in compound 2 and 3, and explains why these compounds can be easily isolated from solution in crystalline form whereas compound 1 requires the presence of large K+ or DMA+ cations. Moreover, the solubility of these compounds in water at room temperature is also consistent with the connectivity of the molecule cluster units within the crystal lattice. Compound 1 is easy to dissolve in cold water whereas compound 2 and compound 3 are much less soluble. This solubility profile mirrors the intermolecular interactions since compound 1 is effectively isolated as a monomer, compound 2 as a dimer and compound 3 as a one dimensional chain (see Fig. 6).
 | ||
| Fig. 6 Scheme summarizing the formation of compounds 1–3. All cations and solvent water molecules are omitted for clarity. Polyhedra: WO6; purple spheres: oxygen; blue spheres: Te; orange spheres: W; black dashed lines: inter-molecular interactions based on Te and O. | ||
Electrospray ionisation mass spectrometry
In order to investigate the composition of these polyoxoanions, single crystals of 1–3 were dissolved in water and acetonitrile mixtures (volume ratio 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) and transferred to the gas phase by electrospray ionisation (ESI).16,17 Although the intact clusters were not directly observed, the fragmentation of compounds 1–3 leads to a range of species (see Fig. 7) which can be assigned to a logical series of heteropoly- and isopolyanionic cluster fragments. Descriptions of the key fragments follows but refer to the SI† for a detailed list of assigned m/zenvelopes. Some cluster fragments were particularly prevalent, for example, the heteropentameric {TeW5O18Na}− (m/z 1358.6) and the isohexameric fragment {W6O19Na}− (m/z 1430.6) were observed in spectra of all three compounds, and were of particularly high intensity. However the MS of compound 1 revealed one key fragment: the doubly charged hetero-octameric anion {[(TeO3)W7O25]Na4H4}2− (m/z 979.7) which corresponds to exactly one quarter of the parent compound (see Fig. 8). In addition, one quarter unit of 1 including the {TeIVO2} heteroatom linker: {[(TeO3)(TeO2)W7O25]H4Na4}2− (m/z 1058.6) was identified in the mass spectra of compound 1. Importantly, the identification of {[(TeO3)2(TeO2)W7O25]H9Na(H2O)}2− (m/z 1123.6) which was observed in the fragmentation of both compounds 2 and 3, corresponds to one quarter of the parent compounds plus the {TeIVO2} heteroatom linker and the pendant {TeIVO3} (see Fig. 7). These observations give important insights to the self-assembly and building block principles developed, establishing the idea that the clusters are constructed from four main building blocks whereby the hetero-anion is involved as a template-linker in the assembly of the cluster squares.
:50) and transferred to the gas phase by electrospray ionisation (ESI).16,17 Although the intact clusters were not directly observed, the fragmentation of compounds 1–3 leads to a range of species (see Fig. 7) which can be assigned to a logical series of heteropoly- and isopolyanionic cluster fragments. Descriptions of the key fragments follows but refer to the SI† for a detailed list of assigned m/zenvelopes. Some cluster fragments were particularly prevalent, for example, the heteropentameric {TeW5O18Na}− (m/z 1358.6) and the isohexameric fragment {W6O19Na}− (m/z 1430.6) were observed in spectra of all three compounds, and were of particularly high intensity. However the MS of compound 1 revealed one key fragment: the doubly charged hetero-octameric anion {[(TeO3)W7O25]Na4H4}2− (m/z 979.7) which corresponds to exactly one quarter of the parent compound (see Fig. 8). In addition, one quarter unit of 1 including the {TeIVO2} heteroatom linker: {[(TeO3)(TeO2)W7O25]H4Na4}2− (m/z 1058.6) was identified in the mass spectra of compound 1. Importantly, the identification of {[(TeO3)2(TeO2)W7O25]H9Na(H2O)}2− (m/z 1123.6) which was observed in the fragmentation of both compounds 2 and 3, corresponds to one quarter of the parent compounds plus the {TeIVO2} heteroatom linker and the pendant {TeIVO3} (see Fig. 7). These observations give important insights to the self-assembly and building block principles developed, establishing the idea that the clusters are constructed from four main building blocks whereby the hetero-anion is involved as a template-linker in the assembly of the cluster squares.
![Mass Spectra data of (C2H8N)6Na18[W28Te8O112]·53H2O (1) (shown in red) and Na28[W28Te10O118]·45H2O (3) (shown in black) in a 50 : 50 water: acetonitrile mixture, transferred to the gas phase by electrospray ionisation (ESI). These spectra show the presence of key ions in the gas phase; in particular: {[(TeO3)W3O9]Na}− (m/z 894.7); {[W4O13]Na}− (m/z 966.8); {[(TeO3)W7O25]H4Na4}2− (m/z 979.7); {[(TeO3)W5O15]Na}− (m/z 1358.6) and {[W6O19]Na}− (m/z 1430.6). The most important features relate to the presence of one quarter unit of 1 including (a) the TeO2 linker: {[(TeO3)(TeO2)W7O25]H4Na4}2− (m/z 1058.6); and (b) one quarter unit of 3 including the TeO2 linker and pendant TeO3: {[(TeO3)2(TeO2)W7O25]H9Na(H2O)}2− (m/z 1123.6). In both (a) and (b) the green mass spectra represent the simulated m/zenvelopes for the reported formulae. It should be noted that the ESI-MS of Na26[W28Te9O115]·37H2O (2) is the same as that of Na28[W28Te10O118]·45H2O (3).](/image/article/2011/SC/c1sc00150g/c1sc00150g-f7.gif) | ||
| Fig. 7
Mass Spectra data of (C2H8N)6Na18[W28Te8O112]·53H2O (1) (shown in red) and Na28[W28Te10O118]·45H2O (3) (shown in black) in a 50:50 water: acetonitrile mixture, transferred to the gas phase by electrospray ionisation (ESI). These spectra show the presence of key ions in the gas phase; in particular: {[(TeO3)W3O9]Na}− (m/z 894.7); {[W4O13]Na}− (m/z 966.8); {[(TeO3)W7O25]H4Na4}2− (m/z 979.7); {[(TeO3)W5O15]Na}− (m/z 1358.6) and {[W6O19]Na}− (m/z 1430.6). The most important features relate to the presence of one quarter unit of 1 including (a) the TeO2 linker: {[(TeO3)(TeO2)W7O25]H4Na4}2− (m/z 1058.6); and (b) one quarter unit of 3 including the TeO2 linker and pendant TeO3: {[(TeO3)2(TeO2)W7O25]H9Na(H2O)}2− (m/z 1123.6). In both (a) and (b) the green mass spectra represent the simulated m/zenvelopes for the reported formulae. It should be noted that the ESI-MS of Na26[W28Te9O115]·37H2O (2) is the same as that of Na28[W28Te10O118]·45H2O (3). | ||
![Hetero-octameric fragment {[(TeO3)W7O25]Na4H4}2− (m/z 979.7) observed in the fragmentation of (C2H8N)6Na18[W28Te8O112]·53H2O (1) in a 50 : 50 water: acetonitrile mixture, which corresponds to one quarter unit of 1. This fragment was observed for all three compounds. Black line is observed spectra and the green line the simulated m/zenvelope.](/image/article/2011/SC/c1sc00150g/c1sc00150g-f8.gif) | ||
| Fig. 8 Hetero-octameric fragment {[(TeO3)W7O25]Na4H4}2− (m/z 979.7) observed in the fragmentation of (C2H8N)6Na18[W28Te8O112]·53H2O (1) in a 50:50 water: acetonitrile mixture, which corresponds to one quarter unit of 1. This fragment was observed for all three compounds. Black line is observed spectra and the green line the simulated m/zenvelope. | ||
The identification of the {TeIVO2} heteroatom linker is commensurate with the X-ray structural analysis since the four {TeIVO4} tetrahedrons link each of the four {W7O25(TeO3)} fragments together, forming a square in the same plane. Such an arrangement may be relatively fragile due to the ring strain in the cluster and thus explains why only the key building block, and not the intact cluster, can be observed using ESI-MS. This hypothesis can be further substantiated by comparing the {O–Te–O} angles between the {TeO3} templates and {TeO4} linkers. Thus, the observation of the quarter fragments in compound 1, and quarter fragments plus linkers and pendant heteroanion in compounds 2 and 3 in the ESI-MS studies are of great relevance to our studies. In summary the mass spectroscopic data clearly show the presence of the quarter unit in solution for compound 1, as well as the Te-linker unit. In addition to the Te-based template and linker units, the pendant Te-units for compounds 2 and 3 have also been identified, which are key for the intra-molecular packing of the clusters in the solid state.
Tube growth
During the course of our recent research into POMs we have discovered that micron-scale tubes (1–100 μm diameter) could be spontaneously grown from a POM crystal which has a poor solubility in water upon addition of a giant organic cation.18,19 Since the tubes produced are both mechanically robust and highly controllable, they might be applied to a wide range of areas such as catalysis, electronics or micro-fluidics.20 Given these features, we opted to explore the possibility of growing tubular architectures from the unique class of polyoxotungstate clusters presented in this work.In preliminary studies we found that tubes could be grown from crystals of all three cluster compounds 1–3, and were observed using optical microscopy. Experimentally, tube growth was carried out by depositing 5 mg of single crystalline samples on a 50 × 22 mm cover slide. Following the addition of 0.15 mL of a 10−2 mol L−1 solution of Methylene Blue to the solution in which the crystals were immersed, the slide was transferred on an Olympus IX81 inverse microscope. Optical micrographs were taken over a time period of 20 min, analysed and processed using the Cell® Software. The tubes grew rapidly with little directional stability and had overall diameters recorded within 2–80 micron diameters and several 1000 microns in length size (Fig. 9).
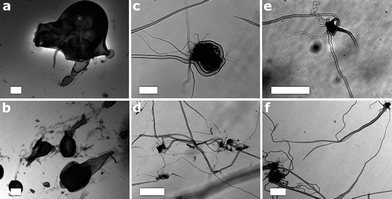 | ||
| Fig. 9 Image of microtubes grown from: (a) and (b) crystals of 1; (c) and (d) crystals of 2 and (e) and (f) crystals of 3 following the addition of 0.15 mL of a 10−2 mol L−1 solution of Methylene Blue. The wandering pattern of growth arises from the relative high solubility single crystals in the Methylene Blue solution. The white scale bar is 100 microns long. | ||
Our initial research showed that the growth process of the tubes is driven by an osmotic pressure which can be controlled by adjusting various parameters, such as the charge of organic cations and the solubility of crystalline POMs. Given the same type of organic cations, if the solubility of POMs is considerable then only very short or even no tubes could be grown. The results of our experiments are consistent with the above conclusions, i.e., compound 1 is more soluble than 2 and 3, thus the growth of long tubes was not observed; while in the case of compounds 2 and 3, long and clear tubes were obtained. More interestingly, the influence of the solubility of a given POM can influence the size and extent of tube growth, for example tubes grown from 1 have the largest diameter; while those from 3 have the smallest.
Conclusions
In summary, a new family in the rare class of macrocyclic heteropolyanions has been discovered by employing a time-dependent synthetic approach. The isolation of this series of nanoscale tellurium-embedded polyoxotungstate cluster ‘squares’ is made possible by refluxing TeIV ions and sodium tungstate in neutral aqueous environment for different time periods. This new class of nanoscale tetrameric tungstotellurite cluster-boxes, (C2H8N)6Na18[W28Te8O112]·53H2O (1), Na26[W28Te9O115]·37H2O (2) and Na28[W28Te10O118]·45H2O (3) have near-identical structures varying only in one or two additional TeO32− units in compound 2 and 3 respectively. Their isolation shows that the TeIV ions in each compound serve not only as templates to the key building units, but also act as linkers between each pair of fragments, thereby enabling the self-assembly of these cyclic structures. Crucially, this means that the Te-based hetero-anions play a key role in directing the primary (TeO3-templated unit), secondary (linking of the TeO3-templated units with TeO4 linkers) and tertiary (aggregation of the clusters by TeO3 ligands to the rims of the clusters) structures of the clusters (where cluster 1 is monomeric, 2 is a dimer and 3 is a 1D chain) defining a totally new type of architectural principle utilizing hetero-anions. Also, by comparing the synthesis of compounds 1, 2 and 3 a time-dependent self-assembly process has been deduced. ESI-MS studies of these materials offer an insight into their possible mechanism of formation which might provide an effective guide to synthesize POMs with new and novel structures rationally in the future. Tube growth from crystals of compounds 1, 2 and 3 demonstrate their ability to form ‘emergent’ materials making them promising candidates to develop new solid state devices. These devices could not only exploit the morphological transformation of the material, but also the intrinsic macrocyclic nature of the building blocks (for ion sensing) along with the acidic and redox properties of HPAs in general (for catalysis) which mean that the tubular architectures will have a range of applications, and could also be deployed on the microfluidic scale.Experimental
Synthesis
It should be noted that the solutions used to form compounds 1–3 can be acidified using either hydrochloric or acetic acid to maintain pH neutral solutions. The reported experimental procedures (above) represent the highest yielding syntheses. Although the chemical formulae may vary slightly as a function of the ions present, the metal-oxide cluster box structures remain the same, despite this change in composition and crystallographic space group alteration.
Measurements
Infrared spectra were measured using samples dispersed in a KBr disk on a Jasco FTIR-410 spectrometer. Wavenumbers are given in cm−1. ICP-OES analysis was carried out at Zentralabteilung für Chemische Analysen, Forschungszentrum Jülich GmbH. Thermogravimetric Analyses (TGA) were performed on a TA Q500 instrument under an atmosphere of air, with heating at a rate of 10.0 °C per minute from room temperature to 600 °C.Electrospray mass spectroscopic measurements
ESI-MS measurements were carried out at 30 °C. The solution of the sample was diluted so the maximum concentration of the cluster ions was of the order of 10−5 M and this was infused at a flow rate of 180 μL h−1. The mass spectrometer used for the measurements was Bruker micro TOF-Q and the data were collected in both positive and negative mode. The spectrometer was previously calibrated with the standard tune mix to give a precision of ca.1.5 ppm in the region of 500–5000 m/z. the standard parameters for a medium mass data acquisition were used and the end plate voltage was set to −500 V and the capillary to +4000 V. The collision cell was set to collision energy to 10 eV with a gas flow rate at 25% of maximum and the collision cell RF was set at 1800 Vpp. Transfer time was set to 60 μs.X-ray crystallography
Suitable single crystals were selected from their respective mother liquors and mounted onto the end of a thin glass fibre using Fomblin oil. X-ray diffraction intensity data were measured at 150(2) K on an Oxford Diffraction Xcalibur Gemini Ultra diffractometer with an ATLAS detector using Cu-Kα radiation [λ = 1.54184 Å]. Structure solution and refinement were performed using SHELXS-9721 and SHELXL-9722via WinGX.23 Corrections for incident and diffracted beam absorption effects were applied using analytical methods.24Acknowledgements
We thank the EPSRC, the Chinese Scholarship Council, WestCHEM and the University of Glasgow for supporting this work and LC thanks the Royal Society/Wolfson Foundation for the award of a merit award.Notes and references
- (a) P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77–112 CrossRef CAS; (b) D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC; (c) D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS; (d) F. Sécheresse, E. Cadot, C. Simonnet-Jegat, in Metal Clusters in Chemistry, (ed.: P. Braunstein, L. A. Oro and P. R. Raithby), Wiley-VCH, Weinheim, Germany, 2008, 1, 124–142 Search PubMed.
- (a) J. T. Rhule, C. L. Hill and D. A. Judd, Chem. Rev., 1998, 98, 327–358 CrossRef CAS; (b) W. B. Kim, T. Voitl, G. J. Rodriguez-Rivera and J. A. Dumesic, Science, 2004, 305, 1280–1283 CrossRef CAS; (c) J. J. Borrás-Almenar, E. Coronado, A. Müller, M. T. Pope, ed. Polyoxometalate Molecular Science; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003 Search PubMed.
- R. Contant and A. Tézé, Inorg. Chem., 1985, 24, 4610–4614 CrossRef CAS.
- (a) S. S. Mal and U. Kortz, Angew. Chem., Int. Ed., 2005, 44, 3777–3780 CrossRef CAS; (b) A. Müller, M. T. Pope, A. M. Todea, H. Bögge, J. van Slageren, M. Dressel, P. Gouzerh, R. Thouvenot, B. Tsukerblat and A. Bell, Angew. Chem., Int. Ed., 2007, 46, 4477–4480 CrossRef; (c) S. G. Mitchell, C. Streb, H. N. Miras, T. Boyd, D.-L. Long and L. Cronin, Nat. Chem., 2010, 2, 308–312 CrossRef CAS.
- M. Leyrie and G. Hervé, Nouv. J. Chemie., 1978, 2, 233–237 Search PubMed.
- M. Bösing, I. Loose, H. Pohlmann and B. Krebs, Chem.–Eur. J., 1997, 3, 1232–1237 CrossRef CAS.
- J. Fischer, L. Ricard and R. Weiss, J. Am. Chem. Soc., 1976, 98, 3050–3052 CrossRef CAS.
- (a) D.-L. Long, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2004, 43, 1817–1820 CrossRef CAS; (b) C. Baffert, J. F. Boas, A. M. Bond, P. Kögerler, D.-L. Long, J. R. Pilbrow and L. Cronin, Chem.–Eur. J., 2006, 12, 8472–8483 CrossRef CAS.
- D.-L. Long, H. Abbas, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2005, 44, 3415–3419 CrossRef CAS.
- J. Yan, D.-L. Long, E. F. Wilson and L. Cronin, Angew. Chem., Int. Ed., 2009, 48, 4376–4380 CrossRef CAS.
- J. Yan, J. Gao, D.-L. Long, H. N. Miras and L. Cronin, J. Am. Chem. Soc., 2010, 132, 11410–11411 CrossRef CAS.
- (a) Crystal data for compound (C2H8N)6Na18[W28Te8O112]·53H2O 1: orthorhombic, Pmmn, a = 21.1146(6) Å, b = 32.0756(10) Å, c = 12.6836(4) Å, V = 8590.1(5) Å3, Z = 2, T = 150 K, 20760 reflections measured, 7091 unique (Rint = 0.0583) which were used in all calculations. Final R1 = 0.0475 and wR2 = 0.1317 (all data); (b) Crystal data for compound Na26[W28Te9O115]·37H2O 2: triclinic, P-1, a = 19.3418(8) Å, b = 21.0639(10) Å, c = 22.8563(11) Å, α = 104.296(4)°, β = 91.843(4)°, γ = 92.426(4)°, V = 9006.8(7) Å3, Z = 2, T = 150 K, 41008 reflections measured, 19584 unique (Rint = 0.0679) which were used in all calculations. Final R1 = 0.0719 and wR2 = 0.2026 (all data); (c) Crystal data for compound Na28[W28Te10O118]·45H2O 3: monoclinic, C2/c, a = 34.8730(1) Å, b = 18.6557(3) Å, c = 27.5438(8) Å, β = 105.384(4)°, V = 17277.4(6) Å3, Z = 4, T = 150 K, 56442 reflections measured, 16202 unique (Rint = 0.0631) which were used in all calculations. Final R1 = 0.0485 and wR2 = 0.1273 (all data).
- J. Yan, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 4117–4120 Search PubMed.
- P. M. Almond and T. E. A. Schmitt, Inorg. Chem., 2002, 41, 5495–5501 CrossRef CAS.
- B. Gao, S. X. Liu, L. H. Xie, X. H. Wang, C. D. Zhang, C. Y. Sun, N. H. Hu and H. Q. Jia, J. Solid State Chem., 2005, 178, 1825–1829 Search PubMed.
- (a) M. T. Ma, T. Waters, K. Beyer, R. Palamarczuk, P. J. S. Richardt, R. A. J. O'Hair and A. G. Wedd, Inorg. Chem., 2009, 48, 598–606 CrossRef CAS; (b) E. F. Wilson, H. N. Miras, M. H. Rosnes and L. Cronin, Angew. Chem., Int. Ed., 2011, 50, 3720–3724 Search PubMed; (c) H. N. Miras, E. F. Wilson and L. Cronin, Chem. Commun., 2009, 1297–1311 RSC.
- (a) T. Waters, X. Huang, X.-B. Wang, H.-K. Woo, R. A. J. O'Hair, A. G. Wedd and L.-S. Wang, J. Phys. Chem. A, 2006, 110, 10737–10741 Search PubMed; (b) H.-J. Zhai, X. Huang, T. Waters, X.-B. Wang, R. A. J. O'Hair, A. G. Wedd and L.-S. Wang, J. Phys. Chem. A, 2005, 109, 10512–10520 CrossRef CAS; (c) L.-S. Wang and X.-B. Wang, J. Phys. Chem. A, 2000, 104, 1978–1990 CrossRef CAS.
- C. Ritchie, G. J. T. Cooper, Y.-F. Song, C. Streb, H. Yin, A. D. C. Parenty, D. A. MacLaren and L. Cronin, Nature Chem., 2009, 1, 47–52 CrossRef CAS.
- G. J. T. Cooper and L. Cronin, J. Am. Chem. Soc., 2009, 131, 8368–8369 CrossRef CAS.
- G. J. T. Cooper, A. G. Boulay, P. J. Kitson, C. Ritchie, C. J. Richmond, J. Thiel, D. Gabb, R. Eadie, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2011, 133, 5947–5954 Search PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Cryst., 2008, A64, 112–122 CrossRef CAS.
- P. Müller, “Crystal Structure Refinement: A Crystallographer's Guide to SHELX”, Oxford, New York, 2006 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef.
- R. C. Clark and J. S. Reid, Acta Cryst., 1995, A51, 887–897 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data and structure refinement details for compounds 1, 2 and 3 and inter-molecular interactions existing in 2 and 3. TGA, ESI-MS and CV experimental details, figures and results. CCDC reference numbers 817467–817471. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00150g |
| This journal is © The Royal Society of Chemistry 2011 |